
一个甲基丙二酸血症家系致病基因的遗传学研究
2022-05-21 04:04邢海星张建芳冯云云
山西医科大学学报 2022年4期
詹 瑛,王 斌,王 宏,邢海星,张建芳,冯云云
(1解放军63750部队医院妇产科,西安 710043;2空军军医大学第一附属医院妇产科;3西安市大兴医院妇产科;*通讯作者,E-mail:zhzhhao@163.com)
甲基丙二酸血症(methylmalonic acidemia,MMA)是由于甲基丙二酰辅酶A变位酶(methylmalonyl-CoA mutase,MCM)自身缺陷或其辅酶腺苷钴胺素(Ado-Cbl)代谢缺陷而导致甲基丙二酸/丙酸/甲基柠檬酸等代谢产物异常堆积而引起的疾病,致死率、致残率高,是我国最常见的有机酸代谢障碍性疾病[1]。我国新生儿发病率为1/26 000~1/3 920,是一种常染色体隐性遗传病[2]。与甲基丙二酸血症相关的基因有14个,其中比较常见的有MUT、MMAA、MMAB、MMADHC、MMACHC、LMBRD1、SLC22A5等,我国较常见的类型为MMACHC基因变异导致的Cb1C型和MUT基因变异导致的单纯性甲基丙二酸血症[3]。MMA患儿临床可表现为嗜睡、呕吐、抽搐、肌张力低下、运动及智力障碍等。MMA临床表型异质性强,临床表现严重程度不一,可病情危重甚至是死亡,也可以仅有轻微甚至无任何症状,及时进行准确的相关致病基因检测对MMA患者的诊断、治疗和预后尤为重要[4]。
本研究基于二代测序(next generation sequencing,NGS)采用目标序列靶向捕获测序技术中最常用的Panel技术对一个甲基丙二酸血症家系进行MMA相关致病基因检测,再结合PCR和Sanger测序在该家系中进行验证,从遗传学的角度初步明确了该家系中先证者MMA的可能发病原因,为其临床诊断提供了有力的证据,为该家系的遗传咨询和产前诊断提供了可靠的分子依据。
1 资料与方法
1.1 临床资料
患儿为2018年12月西安市儿童医院新生儿科收治的1例足月产男婴,临床表现为出生后气促、呻吟不安、肌张力低下、喂养困难。患儿父母双方非近亲结婚,否认家族中遗传病史。曾于2016年7月在陕西省富平县妇幼保健院足月顺产1女,出生后表现为喂养困难、气促、肌张力差,新生儿科诊断为“甲基丙二酸血症”,建议转往上一级医院新生儿科进一步诊疗,因患儿家庭客观原因放弃治疗后患儿于10日龄时夭折。2018年12月该家系母亲足月顺产1男婴,临床表现与前一胎患儿相似,遂紧急转往西安市儿童医院进一步诊疗。患儿入院后化验指标显示酸中毒PCO224 mmHg,HCO3-5.8 mmol/L,低血糖1.0 mmol/L,高血氨127 μmol/L,高乳酸16 mmol/L。血气相色谱-质谱联合有机酸分析结果显示丙酰基肉碱/乙酰基肉碱比值升高,诊断为甲基丙二酸血症,为明确病因遂留取患儿及父母外周血进行基因检测。
1.2 方法
1.2.1 基因组DNA提取和Panel检测 该家系夫妻双方签署知情同意书后,采用EDTA抗凝管抽取患儿及其父母双方静脉血各4 ml,按血液基因组DNA提取试剂盒(北京天根生物科技有限公司)提取外周血基因组DNA,-20 ℃保存备用。患儿DNA样本送至北京康旭医学检验所,利用Sure Select Target Enrichment System目标序列富集试剂盒构建文库,筛选出574个遗传代谢病相关基因制定遗传代谢病目标基因捕获试剂盒捕获外显子及部分内含子区域,应用NEXTSEQ 500测序平台进行二代测序,数据经过质量控制后与基因组参考序列进行比对,结合该家系MMA的临床表型、遗传方式等查阅相关数据库综合分析判断可疑变异的致病性。
1.2.2 PCR和Sanger测序在家系中进行基因变异的共分离验证 根据Panel检测的结果,用PCR结合Sanger测序对患儿及其父母进行突变检测和验证。针对MUT基因的c.753+1delinsTGGTTATT和c.911+1G>C位点采用Primer5.0软件设计跨越2号、3号外显子上下游的引物序列。PCR反应条件为:95 ℃ 10 min,95 ℃ 30 s,60 ℃ 30 s,72 ℃ 45 s,共35个循环,最后72 ℃延伸5 min。PCR产物用1%琼脂糖凝胶电泳检测,之后送上海生工生物工程有限公司进行双向测序。
1.2.3 反转录PCR验证MUT基因突变致病性 患儿外周血细胞RNA的提取采用TianGen RNA提取试剂盒(TianGen,DP419),使用Prime-ScriptTM试剂(Takara Dalian,RR036A)将提取的总RNA进行反转录,依据MUT基因的转录本设计引物进行PCR,MUT-2F:GTGGATACCATATGCAGGAAGC,R:GCACAGTGGATCCCAAAACATT;MUT-5F:AAGAGGCACTAAACACTG,R:CTACCCTACTAGGAAAGC。总RNA进行反转录,扩增产物送上海生工生物工程有限公司测序。测序结果通过与美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)、国际公共数据库(ClinVar)及在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man, OMIM)等公共数据库进行比对分析判断其致病性。
2 结果
2.1 MMA家系可疑致病基因及位点的确定
患儿基因组DNA经Panel检测发现其MUT基因存在c.753+1delinsTGGTTATT变异和c.911+1G>C变异的两处复合杂合突变,两处变异在人群数据库NCBI、OMIM数据库、ClinVar数据库中均未见报道(见图1)。查阅ClinVar与OMIM数据库MUT基因与甲基丙二酸血症相关。甲基丙二酸血症为常染色体隐性遗传病,上述两处变异均为MUT基因内含子区域的变异,其中c.753+1delinsTGGTTATT位点变异位于MUT基因2号内含子下游,紧邻3号外显子,c.911+1G>C位点的变异位于MUT基因3号内含子+1位,均处于“经典剪接区域”,该区域的碱基突变一般均会对原剪切位点产生影响,高度怀疑会影响转录水平内含子序列的剪切,进而影响蛋白质功能。
2.2 Sanger测序验证结果
Sanger测序结果显示患病胎儿MUT基因c.753+1delinsTGGTTATT变异和c.911+1G>C变异分别来自该家系中的母亲及父亲,其父母分别为上述杂合突变的携带者,该遗传方式符合常染色体隐性遗传(见图2),如果上述两处变异为致病性突变,该家系再次妊娠有1/4的概率生育患者。
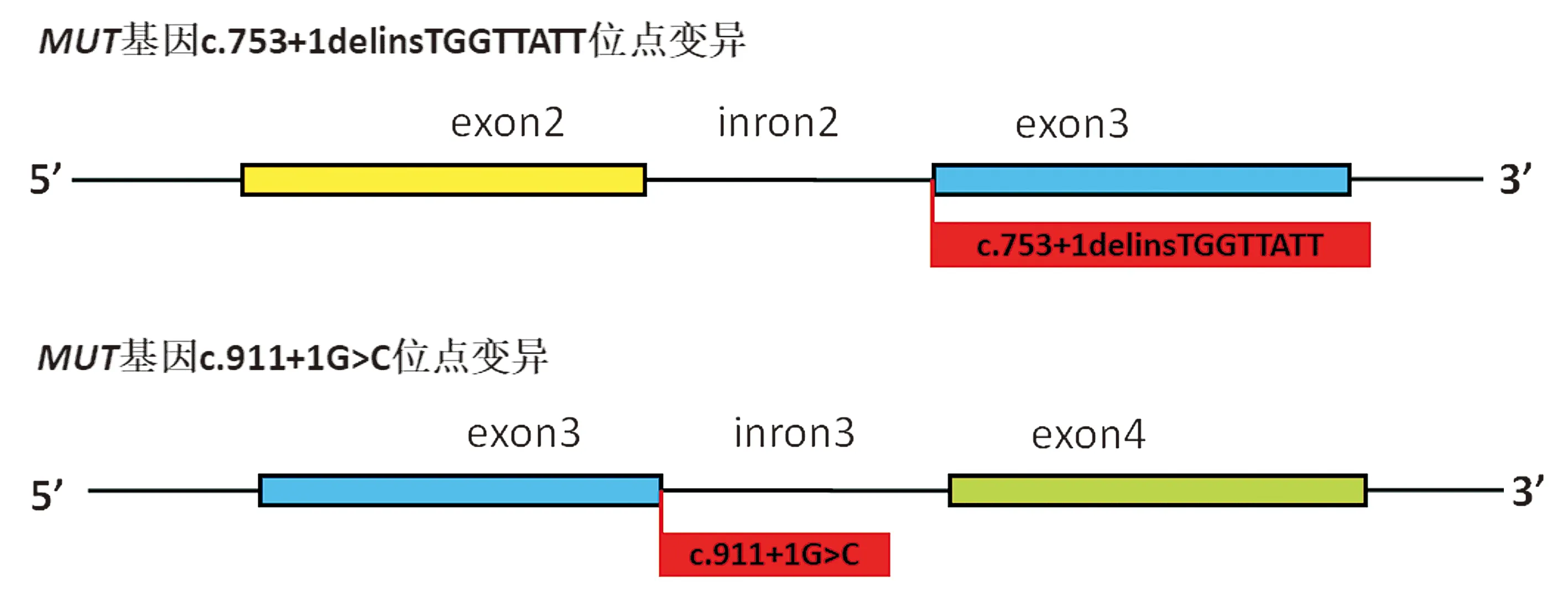
图1 MMA家系患儿MUT基因变异位点在基因序列中的位置Figure 1 The location of the fetal mutations of MUT gene in a family with MMA
2.3 基因突变导致转录后mRNA异常的结果
将患儿总RNA进行反转录PCR(reverse transcription PCR,RT-PCR),测序结果提示该患儿MUT基因的复合杂合突变使得转录过程异常,出现多种异常的转录本。测序结果表明其中一种转录本缺失了3号外显子c.679-c.753这一段碱基序列;另一种转录本缺失了3号外显子c.546-c.753序列以及4号外显子c.754-c.779序列的碱基,同时缺失的碱基替换为4号外显子序列(见图3)。

图3 胎儿MUT基因突变后mRNA水平的变化Figure 3 Changes of mRNA level in the fetus after mutation of MUT gene
3 讨论
甲基丙二酸血症是一种常染色体隐性遗传方式的有机酸代谢性疾病,它的致病基因及临床表型具有高度的异质性,MMA临床症状主要为代谢性酸中毒,拒食、呼吸困难及生长发育迟缓等可以导致新生儿早期死亡或儿童期危重的神经功能障碍[4],临床危害严重,本研究中的患儿最终因严重的酸中毒而死亡。目前该病在全国各地区的发病情况不明,陕西省报道该地区疾病发生率为1/6 960,而其他地区发病率略有升高[5-7]。国内MMA患者60%~80%是以MMACHA基因突变引起的合并型MMA,cblC缺陷是其主要病因,而MUT基因引起的单纯型MMA居于次位[2,8]。
MUT基因定位于6p21,全长约35 kb,内含13个外显子,编码甲基丙二酰辅酶A变位酶MCM[9],该酶是由两个相同亚基组成的同源二聚体,包含两个主要的功能结合区:底物结合区和钴胺素结合区,两个功能区之前存在连接区,MUT基因变异引起氨基酸序列的变化导致MCM酶蛋白功能异常均可能导致单纯型MMA疾病的发生。本研究通过对一个经血液、血气相色谱/质谱串联检查高度怀疑为甲基丙二酸血症的新生儿家系进行了Panel检测,发现患儿患单纯型MMA,患儿MUT基因存在c.753+1delinsTGGTTATT和c.911+1G>C的复合杂合变异,此两处变异均为剪切变异,Sanger测序验证结果显示家系中存在遗传共分离现象,MUT基因很可能为该MMA家系的致病基因。截止2021年10月,ClinVar数据库目前已经报道的MUT基因致病或可能致病的变异位点有460个(https://www.ncbi.nlm.nih.gov/clinvar/),而本研究发现的两处可疑致病位点尚无文献报道。国内学者王斐等[10]对甲基丙二酸血症患儿的MUT基因突变进行统计研究,突变频率较高的位点为c.323G>A、c.729-730insTT与c.1630-1631GG>TA,且MUT基因的变异方式以错义突变多见。而本研究中患儿MUT基因是两处剪切突变的复合杂合突变,经过反转录PCR分析,我们发现这两处剪切突变导致患儿MUT基因的转录本出现异常,其中一种转录本缺失了3号外显子c.679-c.753这一段碱基序列;另一种转录本缺失了3号外显子c.546-c.753序列以及4号外显子c.754-c.779序列的碱基,同时缺失碱基替换为4号外显子序列,这就显著地影响了氨基酸的序列,进而影响到MCM蛋白的功能。已经有研究证实c.755dupA可以导致甲基丙二酸血症[11],说明MUT基因这一碱基附近的变异对蛋白功能影响很大,而本研究中的患儿也存在类似碱基序列的异常。
对于MMA患者家系来讲如何实现疾病的阻断、获得健康的子代是最迫切的需求,实现这一目标的关键在于精准的基因诊断,从而为产前诊断或是胚胎植入前检测提供准确、可靠的分子依据,这就依赖于基因检测技术。目前临床中广泛应用的基因检测技术有一代测序(Sanger测序)、二代测序技术(高通量测序技术)。Sanger测序因为其测序读长长、准确性高的优势,依然被用作其他测序技术验证的金标准,而高通量测序技术以其高通量、自动化程度高、经济成本低的显著特征[12],使遗传学领域的研究以前所未有的速度向前推进。高通量测序技术结合微阵列技术(芯片技术)衍生出了一种新的技术,即目标基因靶向捕获测序技术(targeting sequencing, TS),目前应用最多的是Panel及全外显子测序(whole exome sequencing,WES)。显然,对于Panel技术而言,它具有数据量低、费用较低、与生物学表型有更好的相关性这些显著的优势,而且使得对检测出的变异更容易做出合理解释,同时也避免了检出一些与疾病不相关的或是无意义的变异,加重受检者的经济负担和精神负担。因此美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)的指南建议对于经过科学研究证实的与疾病明确相关的基因,在临床检测中建议使用Panel技术[13]。同时技术的进步也导致了海量临床意义不明的基因变异,其中很大的一部分就是来自剪切位点的变异[14]。正常的剪接过程是极其复杂的,因此我们对剪接过程的了解不能仅根据原始基因序列预测变异的最终效应。最经典的验证方式是通过使用简单的反转录PCR扩增技术,获得扩大的转录组序列,然后与参考转录本进行比对看其是否产生异常剪切,该技术目前仍广泛应用于临床验证,本研究中便是使用了该种方式进行患儿MUT基因剪切突变的验证,有利地证明了两突变个位点的致病性。也可以使用minigene技术或体外剪切技术对剪切变异在体外进行验证,这些体外验证技术帮助解决了一些实际问题,比如无法获得先证者组织样本的问题。而微阵列技术的发展和RNA-seq技术的改进解决了这一问题,随后出现的高通量测序技术能够实现直接对RNA序列进行测序,但是迄今为止因为其经济成本较高,需要掌握大量的生物信息学数据分析能力,还不能够做到全面普及应用[15]。
总而言之,对曾经生育过MMA患儿的家系采取针对性的可疑致病基因检测策略,可以快速、准确地做出基因诊断,为患儿及家系的诊断和治疗提供可靠的分子依据。科学地在家系中进行可疑致病基因的验证,可以为患者家系下一步实施产前诊断或是胚胎植入前筛查打下坚实的基础。
猜你喜欢
分子催化(2022年1期)2022-11-02
分子诊断与治疗杂志(2022年9期)2022-10-09
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
佛山陶瓷(2018年6期)2018-09-14
森林工程(2018年1期)2018-05-14
湖北农业科学(2014年11期)2014-09-10
中学理科·综合版(2008年11期)2008-01-14
