
Toll样受体7基因敲除对水疱性口炎病毒增殖的影响
2022-06-06 05:26孟洁洁樊文杰邢嘉友褚贝贝杨国宇王梦迪
中国畜牧兽医 2022年6期
孟洁洁,宋 月,樊文杰,杨 乐,邢嘉友,王 江,褚贝贝,杨国宇,王梦迪
(1.河南农业大学,农业农村部动物生化与营养重点开放实验室,郑州 450046;2.河南牧业经济学院,食品与生物工程学院,郑州 450046)
水疱性口炎(vesicular stomatitis,VS)又称烂舌症,是由水疱性口炎病毒(Vesicular stomatitis virus,VSV)引起牛、马、猪等哺乳动物的一种高度接触性传染病,以水疱性病变为特征,可通过摩擦接触破损的动物皮肤引起感染,最早出现在美国[1-2]。VS的流行具有季节性、暴发性的特点,较易在夏季发生,通常可在2 h内侵犯牧场内的大批牲畜,给养殖户带来巨大的经济损失。VSV属于单股负链RNA(single stranded negative stranded RNA,ssRNA)病毒,也是一种能够由昆虫传播的横纹肌病毒,它具有囊膜,并在小鼠中具有神经毒性[3],是由包括狂犬病毒在内的嗜神经病毒组成的弹状病毒家族的原型成员。水疱性口炎是由VSV中的糖蛋白G在病毒吸附、内吞和膜融合过程中介导的[1]。糖蛋白G作为VSV的病原相关分子模式(pathogen-associated molecular patterns,PAMP),可以与Toll样受体7(Toll-like receptor 7,TLR7)相互作用并激活自噬,从而启动抗病毒程序[4-6]。
Toll样受体(Toll-like receptors,TLR)是先天免疫中的模式识别受体(pattern recognition receptors,PRRs),可以通过与配体相互识别被激活,使信号向下游传递,参与适应性免疫应答。TLRs至少有11种,均属于Ⅰ型跨膜蛋白,TLR7也不例外,它由胞外区、跨膜区和胞内区3个部分组成,其中胞外区可与配体相互识别,跨膜区含有较多的半胱氨酸,胞内区与白细胞介素受体同源,负责信号传递。TLR7还能够特异性识别短链RNA及其核苷类似物,在与其配体特异性结合后,通过信号传导最终可以促进Ⅰ型干扰素的表达,参与到抵抗单链RNA病毒感染的天然免疫中[7-11]。本试验利用CRISPR/Cas9基因定点修饰技术构建PK15细胞TLR7基因稳定敲除细胞系,并研究TLR7基因敲除对VSV-GFP毒株复制规律的影响,以期为防控VSV等RNA病毒引起的疾病提供新思路。
1 材料与方法
1.1 材料
猪肾上皮细胞(porcine renal epithelial cells,PK15)、人胚肾上皮细胞衍生细胞系(human embryonic renal epithelial cell derived cell line,HEK293T/17)、非洲绿猴肾上皮细胞(renal epithelial cells of African green monkey,Vero)、VSV-GFP毒株、大肠杆菌Top10感受态细胞均由农业农村部动物生化与营养重点开放实验室保存;LentiCRISPR V2、pMD2.G以及pSPA×2均购自Addgene公司;T7核酸内切酶及T7 NEBuffer均购自NEB公司;一步法反转录试剂盒购自北京密码子生物科技有限公司;实时荧光定量PCR反应试剂盒购自宝生物工程(大连)有限公司;嘌呤霉素、血清、培养基等均购自Thermo公司;CCK-8细胞毒性检测试剂盒购自南京建成生物工程研究所。
1.2 引物设计与合成
根据NCBI数据库公布的猪源TLR7基因组序列(GenBank登录号:NM_001097434.1)进行sgRNA预测,最终设计sgRNA1、sgRNA2、sgRNA3序列用于TLR7基因敲除。此外,在NCBI数据库中查找猪源VSV-N基因的mRNA序列 (GenBank登录号:MK934319.1),并利用Primer Premier 6.0软件设计引物用于实时荧光定量PCR(表1)。引物均由生工生物工程(上海)股份有限公司合成。
1.3 TLR7-sgRNA载体构建
LentiCRISPR v2质粒在BsmBⅠ酶的作用下于37 ℃反应1 h完成单酶切,在电泳并回收后,利用合成的sgRNA引物对分别进行以下反应。引物退火杂交的程序为:95 ℃变性10 min;95 ℃梯度退火至85 ℃(每秒钟降低2 ℃),85 ℃梯度退火至25 ℃(每秒钟降低0.1 ℃);4 ℃保存1 h。在T4 DNA连接酶的作用下,酶切所得大片段与上述产物在4 ℃过夜反应。转化摇菌涂布平板,然后置于37 ℃温箱中过夜生长,挑取形状色泽等良好的单菌落送测序。选取测序无误的菌液进行质粒提取,稀释成1 000 ng/μL。
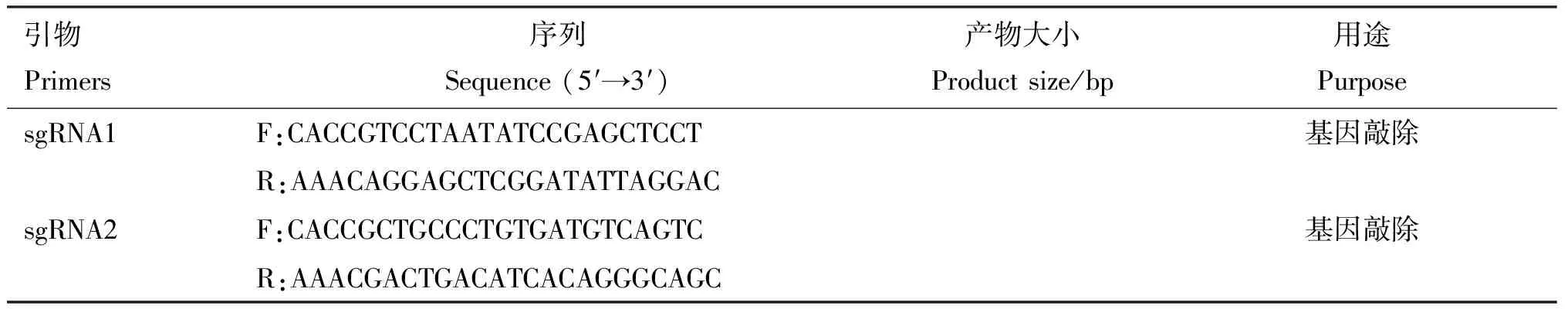
表1 引物信息

续表
1.4 TLR7基因敲除细胞系的构建
1.4.1 慢病毒包装 将HEK293T/17细胞接种于T-25细胞培养瓶,待细胞融合度达到40%左右时进行转染。将包装质粒(pMD2.G)、包膜蛋白质粒(pSPAX2)与构建的重组转移质粒共转入HEK293T/17细胞,于37 ℃、5% CO2培养箱中分别培养48、72 h后收取上清液,将其混合后得到慢病毒液,于-80 ℃保存备用。
1.4.2 多克隆细胞株筛选 将PK15细胞接种于6孔板中,待细胞融合度达到40%左右时进行慢病毒感染。取1 mL慢病毒液混合1 mL DMEM处理PK15细胞。当慢病毒感染细胞48 h后,用含有10% FBS的DMEM培养基(含有8 μg/mL嘌呤霉素)继续培养细胞,当未感染细胞被杀死后,即可留下所需的阳性细胞。以未作处理的PK15细胞为对照,当对照组细胞全部死亡时,将嘌呤霉素浓度更换为3 μg/mL再进行培养,最后将筛选出来的多克隆细胞进行细胞传代培养。
1.4.3 Western blotting检测sgRNA编辑效率 分别接种PK15及筛选出来的3株多克隆细胞于60 mm培养皿中,每皿细胞数为7×105个。待细胞汇合度达到60%时收取目的蛋白,将得到的蛋白进行BCA浓度检测后,取相应量蛋白进行电泳并转移到聚偏二氟乙烯(polyvinylidene fluoride,PVDF)膜上,5%脱脂乳室温封闭1 h后,加入TLR7单克隆抗体(1∶1 000稀释)过夜(4 ℃),利用1×TBST洗膜3次后,用山羊抗兔IgG(1∶5 000稀释)孵育1 h,利用1×TBST洗膜3次后进行显影,所选用内参蛋白为β-actin。
1.4.4 单克隆细胞系的筛选 选取编辑效率高的多克隆细胞,通过有限稀释法进行单克隆细胞系的筛选。再分别将PK15细胞与所得的PK15-TLR7-/-单克隆细胞系接种于60 mm培养皿中,按照1.4.3方法收取蛋白并进行Western blotting检测。根据检测结果,进一步提取PK15-TLR7-/-单克隆细胞系基因组并进行PCR扩增,将产物送至生工生物工程(上海)股份有限公司进行测序。
1.5 细胞形态观察
将PK15、PK15-TLR7-/-细胞接种于100 mm培养皿中,于0、6、12、24、36、48 h在光学显微镜下仔细观察细胞形态、大小以及密集程度并拍照。
1.6 CCK-8检测
将PK15、PK15-TLR7-/-细胞接种于96孔板中,分别于0、6、12、24、36、48 h进行CCK-8检测,每个时间点做3个重复,加入CCK-8处理后在37 ℃、5% CO2培养箱中孵育2 h,分别检测2种细胞在450 nm处的吸光度值。
1.7 流式细胞仪检测
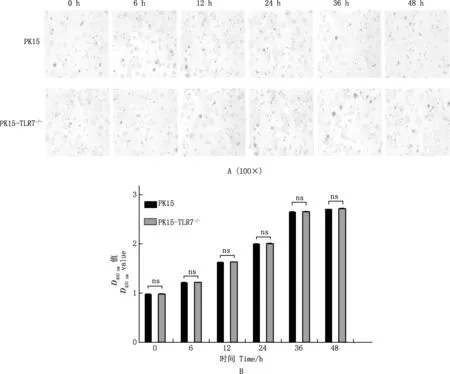
将PK15、PK15-TLR7-/-细胞接种于24孔板中,当细胞汇合度达到50%时进行细胞计数,采用VSV-GFP感染细胞,病毒感染复数(multiplicity of infection,MOI)为0.01。将计算好的病毒量加入DMEM,混合的病毒悬液涡旋8 s后处理细胞,于37 ℃吸附 1 h后弃去原培养基,PBS清洗后再加入新鲜含有2% FBS的DMEM培养基,分别于0、6、12、24、36 h观察细胞形态及荧光情况并记录,同时按照时间点收样并进行流式细胞仪检测。
1.8 实时荧光定量PCR检测
将PK15、PK15-TLR7-/-细胞接种于35 mm培养皿中,待细胞融合度达到50%时进行细胞计数,利用VSV-GFP感染细胞,MOI为0.01。按照1.7方法进行处理后,分别于0、2、4、6、12、24、36 h进行收样,先加入Trizol进行总RNA提取。获得总RNA后进行反转录合成cDNA,利用设计的实时荧光定量PCR引物进行扩增。PCR反应体系10 μL:SYBR Premix ExTaqⅡ 5 μL,上、下游引物(20 μmol/L)各0.4 μL,cDNA 1.5 μL,ddH2O 2.7 μL。PCR反应程序:95 ℃预变性2 min;95 ℃变性20 s,60 ℃退火20 s,72 ℃延伸20 s,共40个循环;绘制熔解曲线,25 ℃反应1 min。
1.9 Western blotting 检测VSV-GFP的GFP蛋白的表达
分别将PK15、PK15-TLR7-/-细胞接种于60 mm培养皿中。待细胞汇合度达到60%时进行细胞计数,利用VSV-GFP感染细胞,MOI为0.01。按照1.7方法进行处理后,分别于0、4、6、12、24、36 h收样并提取目的蛋白。一抗为GFP单克隆抗体(1∶1 000稀释),二抗为山羊抗鼠IgG(1∶5 000),洗膜液为1×TBST,内参蛋白为β-actin,方法同1.4.3。
1.10 VSV-GFP子代病毒滴度测定
分别将PK15、PK15-TLR7-/-细胞接种于6孔板中,待细胞汇合度达到50%时进行细胞计数后,以VSV-GFP感染细胞,MOI为0.01。按照1.7方法进行处理后,分别于0、4、6、12、24、36 h进行收样,将样品反复冻融且每次冻存时间不少于8 h,冻融3次后在超净工作台内吸取上清吹打底部的细胞碎片,离心后留取上清液。在96孔板中接种Vero细胞,设置12个病毒浓度梯度,以10倍为一个浓度差进行倍比稀释后处理细胞,于37 ℃、5% CO2培养箱中吸附1 h,弃去培养基,用1×PBS清洗2次,换成含有2% FBS的DMEM培养基进行培养。每隔1 d观察1次,5~6 d后,观察细胞病变情况,记录病变孔并计算半数组织培养感染剂量(TCID50),最终得到VSV-GFP子代病毒滴度变化。
1.11 数据统计分析
每种试验均重复3次,利用Excel软件整理数据并进行双尾T检验分析,数据以平均值±标准差表示,利用GraphPad Prism 8.3软件进行统计学分析并作图,P<0.05表示差异显著,P<0.01表示差异极显著。
2 结 果
2.1 载体鉴定结果
用BsmBⅠ对LentiCRISPR v2载体单酶切后进行电泳检测,以LentiCRISPR v2质粒作为对照,结果显示该载体单酶切后有13 000和1 873 bp 2个目的条带(图1A),与预期一致。 测序结果表明成功构建了重组质粒pTLR7-sgRNA1、pTLR7-sgRNA2和pTLR7-sgRNA3(图1B)。

A,LentiCRISPR v2酶切电泳图:M,DL15000 DNA Marker;1,LentiCRISPR v2质粒;2,LentiCRISPR v2单酶切产物。B,序列测定结果A,Enzyme-digestion electrophoresis of LentiCRISPR v2:M,DL15000 DNA Marker;1,Plasmid LentiCRISPR v2;2,Results of LentiCRISPR v2 enzyme digestion.B,Sequence determination results图1 Cas9/sgRNA载体的构建Fig.1 Construction of the Cas9/sgRNA vector
2.2 PK15-TLR7-/-单克隆细胞系的构建
Western blotting检测3个sgRNA构建的PK15-TLR7-/-多克隆细胞,结果表明设计的sgRNA1、sgRNA2、sgRNA3均可对TLR7基因进行有效编辑,其中编辑效率最高的为sgRNA2(图2A)。将编辑效率最高的sgRNA2构建的PK15-TLR7-/-多克隆细胞所筛选得到的单克隆细胞系进行Western blotting检测,结果显示,无TLR7蛋白表达(图2B)。测序结果表明,与正常TLR7基因相比,2号靶位点处插入碱基A导致后续碱基发生移码突变,证明PK15-TLR7-/-敲除细胞构建成功(图2C)。

A,Western blotting检测sgRNA编辑效率:1、2,分别为PK15细胞、sgcontrol细胞;3~5,分别为用sgRNA1、sgRNA2、sgRNA3构建的PK15-TLR7-/-多克隆细胞。B,Western blotting检测PK15细胞和PK15-TLR7-/-单克隆细胞中TLR7蛋白的表达情况。C,PK15-TLR7-/-单克隆细胞测序结果A,Western Blotting results of sgRNA editing efficiency detection:1 and 2,PK15 cells and sgcontrol cells,respectively;3-5,PK15-TLR7-/- polyclonal cells constructed with sgRNA1,sgRNA2 and sgRNA3,respectively.B,Western blotting results of TLR7 protein expression in PK15 cells and PK15-TLR7-/- monoclonal cells detection.C,Results of PK15-TLR7-/- monoclonal cell sequencing图2 单克隆细胞系的构建Fig.2 Construction of monoclonal cell lines
2.3 TLR7基因敲除对PK15细胞增殖和形态的影响
通过光学显微镜在不同时间点观察PK15与PK15-TLR7-/-的细胞形态发现,敲除TLR7基因并不影响PK15的细胞形态(图3A)。利用CCK-8检测敲除TLR7基因对PK15细胞增殖的影响发现,PK15-TLR7-/-细胞的活力与PK15细胞无显著差异(P>0.05)(图3B)。
2.4 PK15细胞中TLR7基因敲除对VSV-GFP复制的影响
在VSV-GFP感染PK15细胞与PK15-TLR7-/-细胞后的不同时间点,通过荧光显微镜进行荧光检测发现,随着时间增加,荧光强度逐次增加,且PK15-TLR7-/-细胞的GFP荧光强度强于PK15细胞(图4A);与此同时,流式细胞术检测结果显示,相同时间点的PK15-TLR7-/-细胞感染VSV-GFP的比例显著或极显著高于PK15细胞(P<0.05;P<0.01)(图4B和4C),表明敲除TLR7基因促进了VSV-GFP在PK15细胞中的复制。
2.5 PK15细胞中TLR7基因敲除对VSV-N基因mRNA水平的影响
当VSV-GFP感染PK15与PK15-TLR7-/-细胞后,利用实时荧光定量PCR检测不同时间点VSV-N基因mRNA相对表达量发现,随着时间增加,VSV-N基因mRNA相对表达量逐渐增加。感染4~36 h时,PK15细胞中VSV-N基因的mRNA相对表达量显著或极显著低于PK15-TLR7-/-细胞(P<0.05;P<0.01)(图5),表明敲除PK15细胞中TLR7基因能显著促进VSV-N基因表达。
2.6 PK15细胞中TLR7基因敲除对VSV-GFP GFP蛋白表达的影响
当VSV-GFP感染PK15与PK15-TLR7-/-细胞后,Western blotting检测不同时间点VSV-GFP GFP蛋白的表达量变化发现,PK15与PK15-TLR7-/-细胞中的VSV-GFP GFP蛋白均从6 h开始表达,并且随着时间的增加,VSV-GFP GFP蛋白的表达量均逐渐增多,但相较于PK15细胞来说,在PK15-TLR7-/-细胞中VSV-GFP GFP蛋白含量更高(图6),因此,在PK15细胞中敲除TLR7基因能够促进VSV病毒的复制。
*,差异显著(P<0.05),**,差异极显著(P<0.01),ns,差异不显著(P>0.05)。下同*,Significant difference (P <0.05),**,Extremely significant difference (P<0.01),ns,No significant difference (P>0.05).The same as below图3 PK15与PK15-TLR7-/-细胞的形态(A)与活力(B)Fig.3 Morphology (A) and activity (B) of PK15 and PK15-TLR7-/- cells
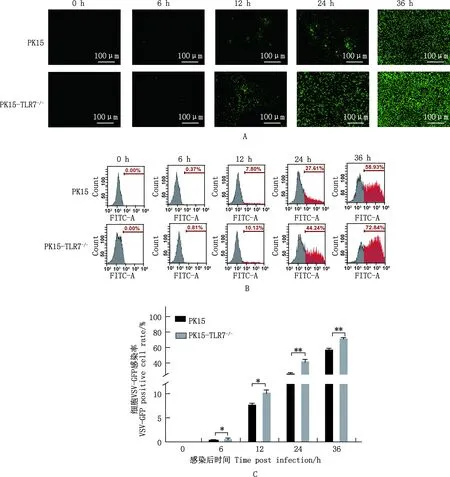
A,荧光显微镜观察结果(40×);B,流式细胞仪检测结果;C,不同时间VSV-GFP的感染率A,Results of fluorescence microscopy (40×);B,The results of flow cytometry;C,Results of infection rate of VSV-GFP at different time图4 TLR7基因敲除对VSV-GFP复制的影响Fig.4 Effect of TLR7 gene knockout on replication of VSV-GFP
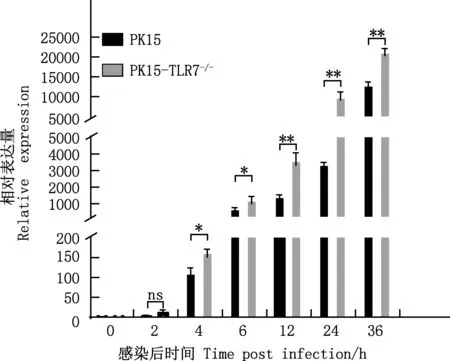
图5 TLR7基因敲除对VSV-N基因mRNA表达水平的影响Fig.5 Effect of TLR7 gene knockout on VSV-N gene mRNA expression
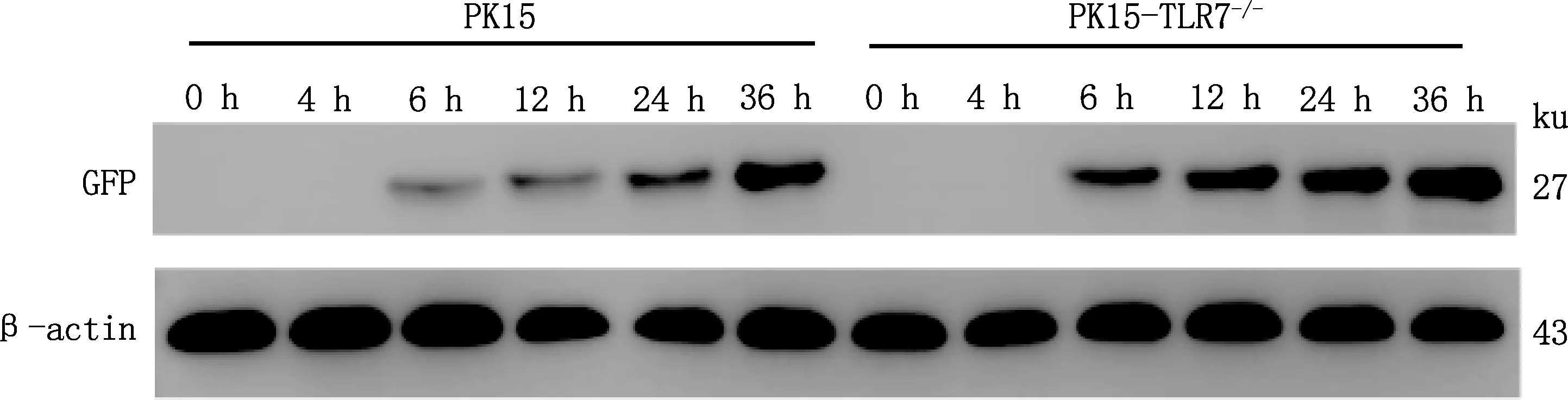
图6 TLR7基因敲除对VSV-GFP GFP蛋白表达的影响Fig.6 Effect of TLR7 gene knockout on VSV-GFP GFP protein expression
2.7 PK15细胞中TLR7基因敲除对VSV-GFP子代病毒滴度的影响
当VSV-GFP感染PK15与PK15-TLR7-/-细胞后,检测不同时间点VSV-GFP子代病毒滴度,结果表明,感染VSV-GFP 6 h后子代病毒开始释放,此时PK15细胞中VSV-GFP子代病毒滴度低于PK15-TLR7-/-细胞,但差异不显著(P>0.05);随着VSV-GFP感染时间的增加,PK15细胞中VSV-GFP子代病毒滴度显著或极显著低于PK15-TLR7-/-细胞(P<0.05;P<0.01)(图7)。说明TLR7基因敲除能够促进VSV-GFP子代病毒的产生。
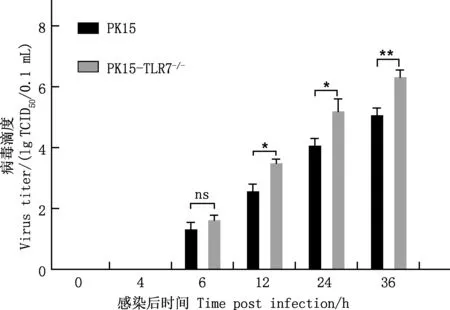
图7 TLR7基因敲除对VSV-GFP子代病毒滴度的影响Fig.7 Effect of TLR7 gene knockout on titer of VSV-GFP progeny virus
3 讨 论
本试验选择猪PK15细胞作为研究对象,并利用CRISPR/Cas9技术获得PK15-TLR7-/-稳定的单克隆细胞系。 CRISPR/Cas9基因编辑技术是一种由RNA指导Cas核酸酶对靶向基因进行特定DNA精准修饰的技术[12],作为ZFN和TALEN后的第3代基因编辑技术,它具有经济快捷、操作简便、效率高等特点,被广泛应用于基因功能的挖掘、动植物品系的培育以及动物模型的建立等研究领域[13-15]。Xu等[16]利用该技术制备了无外源标记的CD163和pAPN双等位基因编辑猪,在不影响猪正常生长发育和繁殖性能的前提下,显著降低了对猪繁殖与呼吸综合征病毒和猪传染性胃肠炎病毒的易感性。Yang等[17]使用CRISPR/Cas9基因编辑技术与体细胞核移植(SCNT)相结合,获得了血红蛋白清道夫受体CD163基因敲除的杜洛克猪,并证明了敲除CD163基因的杜洛克猪可完全抵抗高致病性猪繁殖与呼吸综合征病毒感染。Hübner等[18]利用CRISPR/Cas9基因编辑系统靶向切割ASFV-p30基因组,导致由ASFV引起的空斑完全消失,最终使病毒产量降低了4个数量级。因此,利用CRISPR/Cas9基因编辑技术可以用于研究病毒与宿主细胞的联系、病毒的致病机理等。
TLR7基因于2000年发现[19],它广泛表达于多种细胞的内体-溶酶体中,能够识别某些小分子的抗病毒化合物和病毒mRNA介导的抗病毒的天然免疫反应[19-21]。 当TLR7被活化后,能够招募髓系分化因子88(myeloid differentiation protein 88,MyD88),进而使MyD88活化并招募白细胞介素-1受体相关激酶(interleukin-1 receptor-associated kinase,IRAK),形成4层左手螺旋状信号复合物,使信号向下游传递,最终导致干扰素调节因子(interferon regulatory factor 7,IRF7)磷酸化后入核,产生Ⅰ型干扰素以发挥抗病毒作用[22]。如Martin等[23]通过利用呼吸道合胞病毒感染BALB/c小鼠发现,TLR7的表达量上升,同时释放了细胞因子IRF5;Diebold等[24]用聚乙烯亚胺与浓缩流感病毒ssRNA的复合物(PEI-RNA)活化野生型小鼠骨髓细胞产生IFN-α,但不能活化TLR7-/-小鼠的骨髓细胞。此外,VSV的宿主非常广泛,能够感染植物、无脊椎动物和有脊椎动物,实际生产中也会引起猪急性、热性、高度接触性的传染病[25]。而Lund等[26]用VSV或流感病毒体外感染鼠骨髓细胞,可分泌高水平的IFN-α和白细胞介素-12(interleukin-12,IL-12),而TLR7-/-小鼠来源的骨髓细胞丧失了对VSV的反应,也表明TLR7能够活化免疫细胞,并促进炎性细胞因子的释放,从而发挥抗病毒机制。因此,宿主细胞能够通过TLR7识别病毒的RNA,从而启动TLR7依赖的抗病毒机制。本研究发现,TLR7基因敲除可以促进VSV在PK15细胞中的复制,但不影响PK15细胞的形态及活力,进一步验证了TLR7在宿主细胞抗病毒中扮演着重要的角色。但VSV如何与TLR7相互作用以及TLR7影响病毒复制生活周期中的哪一阶段,仍需进一步研究证明。本研究初步验证了TLR7在天然免疫中的作用,为VSV等RNA病毒性疾病的防控提供新的思路和策略。
4 结 论
本研究成功构建了猪pTLR7-sgRNA重组质粒并最终得到了PK15-TLR7-/-单克隆细胞系,通过检测敲除TLR7基因对VSV-GFP复制情况的影响,证明敲除TLR7基因在一定程度上促进了VSV复制。
猜你喜欢
中学生物学(2022年8期)2022-10-13
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
中国现代医生(2022年21期)2022-08-22
广东教育·职教版(2021年3期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
江苏农业学报(2019年1期)2019-09-10
奥秘(2019年8期)2019-08-28
学校教育研究(2018年27期)2018-05-14
小猕猴学习画刊(2017年3期)2017-07-19
小猕猴智力画刊(2016年6期)2016-05-14
