
Myelinosome organelles in pathological retinas:ubiquitous presence and dual role in ocular proteostasis maintenance
2022-09-27 09:44MarinaYefimova
中国神经再生研究(英文版) 2023年5期
Marina G.Yefimova
Abstract The timely and efficient elimination of aberrant proteins and damaged organelles, formed in response to various genetic and environmental stressors, is a vital need for all cells of the body.Recent lines of evidence point out several non-classical strategies employed by ocular tissues to cope with aberrant constituents generated in the retina and in the retinal pigmented epithelium cells exposed to various stressors.Along with conventional strategies relying upon the intracellular degradation of aberrant constituents through ubiquitin-proteasome and/or lysosome-dependent autophagy proteolysis,two non-conventional mechanisms also contribute to proteostasis maintenance in ocular tissues.An exosome-mediated clearing and a myelinosome-driven secretion mechanism do not require intracellular degradation but provide the export of aberrant constituents and “waste proteins”outside of the cells.The current review is centered on the non-degradative myelinosome-driven secretion mechanism, which operates in the retina of transgenic Huntington’s disease R6/1 model mice.Myelinosome-driven secretion is supported by rare organelles myelinosomes that are detected not only in degenerative Huntington’s disease R6/1 retina but also in various pathological states of the retina and of the retinal pigmented epithelium.The intra-retinal traffic and inter-cellular exchange of myelinosomes was discussed in the context of a dual role of the myelinosome-driven secretion mechanism for proteostasis maintenance in different ocular compartments.Special focus was made on the interplay between degradative and non-degradative strategies in ocular pathophysiology, to delineate potential therapeutic approaches to counteract several vision diseases.
Key Words: autophagy; Huntington’s disease; Müller cells; myelinosome-driven secretion;myelinosomes; ocular pathophysiology; proteostasis; retina; retinal pigmented epithelium; ubiquitinproteasome system
Introduction
Anatomically and developmentally, the retina is an extension of the central nervous system, manifesting the highest oxidative metabolism in the body,a particular polyunsaturated fatty acid composition of cell membranes, and a permanent exposure to light irradiation (Hurley, 2021).Such peculiarities determine the vulnerability of the retina to various genetic, metabolic,and environmental stressors (nutrient deprivation, diabetes, hypertension,hypoxia, high temperature, smoking, etc), which affect cellular proteome composition and proteostasis (Calandria et al., 2012).
The term proteostasis concerns the events restoring the balance of the synthesis, trafficking, localization, aggregation/disaggregation, and degradation of proteins to sustain a dynamic equilibrium of activity that satisfies cell demands in specific situations (Calandria et al., 2012).This is achieved by the action of many proteins, which form a proteostasis network,incorporating translational machinery, chaperones, and proteins involved in the ubiquitin-proteasome (UPS) and autophagy machinery (Paraoan et al., 2020).By classical definition, components that govern stress responses,such as unfolded protein response, are considered supplementary but crucial to the proteostasis network (Labbadia and Morimoto, 2015).UPS and autophagy are two main degradative pathways for the removal of abnormal/aberrant proteins in all cells of the body (Kocaturk and Gozuacik, 2018; Li et al., 2022).The first one involves translocation of the misfolded protein out of the endoplasmic reticulum (ER) and shuttling to the proteasome.The second one is a lysosome-dependent catabolic process in which cellular elements are encapsulated within double-membrane vesicles termed autophagosomes that fuse with lysosomes for degradation.When proteostasis is disrupted, the aberrant proteins accumulate, inducing the formation of toxic subproducts and inclusion bodies/aggregates (Calandria et al., 2012).
Recent lines of evidence point out several non-classical strategies employed by ocular tissues to cope with aberrant proteins generated in the retina and retinal pigmented epithelial (RPE) cells exposed to various genetic and environmental stressors.Along with conventional strategies relying upon the intracellular degradation of aberrant proteins through UPS and/or lysosomedependent autophagy proteolysis, two non-conventional mechanisms have been revealed: an exosome-mediated clearing (Wang et al., 2009) and a myelinosome-driven secretion (MDS) (Yefimova et al., 2021).Neither of which requires the intracellular degradation of abnormal/aberrant proteins,but provides their export outside of the cells.Thus, exosome-mediated clearing ensures the export of “waste” proteins by RPE cells.An imbalance in exosome-mediated clearing leads to the formation of drusen deposits in age-related macular degeneration (ARMD) (Blasiak et al., 2019).By MDS mechanism, the retinal glial Müller cells export mutant Huntingtin (mHTT)protein in the extracellular milieu, maintaining thereby their proteostasis, but contributing to mHTT spreading over the retina of the transgenic Huntington’s disease (HD) model mice (Yefimova et al., 2021).In this review, we will discuss the MDS strategy concerning degenerative states of the retina of various etiologies.
Search Strategy and Selection Criteria
The studies cited in the current review, published from 1938 to 2022,were retrieved by an electronically search on Google, Web of Science, and PubMed databases using the following keywords/terms: myelinosomes,myelin-like, membranous whorls, membranous bodies, multimembranous bodies, membrane swirls, membranous structures, vesiculated membranes,retina, retinitis pigmentosea, drusen, age-related macular degeneration,retinal degeneration, glaucoma, Alzheimer’s disease, RCS rats, autophagy,proteasome, proteostasis, light-induced retinal degeneration, rhodopsin mutation, Huntington’s disease, oxidative stress.Furthermore, we also used various combinations of the above search terms to reach the literature data more specifically.
Non-Conventional Myelinosome-Driven Secretion Strategy in Retinal Degenerations
Myelinosomes were described years ago by electron microscopy investigators as rare intracellular electron-dense organelles, arising in cells exposed to different stress conditions (Ghadially, 1997).In ultra-thin transmission electron microscopy (TEM) transversal sections, myelinosomes show a stacked, whorled or reticulated arrangement, which closely resembles myelin sheath insulating the axons of neurons.This complicates the detection of myelinosomes in micrographs from brain sections, but not from the retinal ones because the axons of retinal neurons are not insulated with myelin sheath (Zhu et al., 2018).An electron-lucid matrix is often discerned in myelinosome organelles in the cross-sections from different tissues.As usual, by TEM technique, myelinosome matrix does not present a welldefined arrangement, while sometimes the honeycomb-like organization is discerned.Various names have been given to these organelles, which are also known as multimembranous bodies, multilamellar bodies, myelin bodies,myelonoid bodies, myelin figures, myelin-like organelles, membranous bodies,vesiculated membranes, or zebra bodies, thereby confusing the identification of myelinosomes in different normal and pathological tissues (Ghadially,1997).The non-conventional MDS strategy mediated by myelinosome organelles will be discussed in this review in the context of degenerative states of the outer and inner retina.
By definition, retinal degenerations are a group of progressive disorders with diverse etiologies leading to a common outcome, which is neuronal cell death.Two common features of retinal degenerations are as follows: cellular inability to manage structurally damaged abnormal proteins and enhanced oxidative stress (Calandria et al., 2012).These features are functionally and causatively interrelated.Thus, when the overproduction of aberrant proteins is sustained and the cells are unable to cope with the imbalance, the system tends toward overproduction of reactive oxygen species (ROS).Reciprocally,oxidative conditions induce protein damage and an impairment of the UPS system, leading to an accumulation of abnormal proteins.Nevertheless,while deleterious for UPS system, the oxidative stress activates autophagy – a second degradative proteostasis strategy, which tends to restore the dynamic equilibrium in the cells (Calandria et al., 2012).
Transgenic Huntington’s Disease Mice
HD is a fatal autosomal dominant late-onset neurological disorder that causes progressive and irreversible motor dysfunctions, resulting in coordination and gait difficulties, as well as cognitive and behavioral changes (McColgan and Tabrizi, 2017).Belonging to the class of repeat expansion diseases (Paulson,2018), HD is caused by the expansion of CAG triplets in the gene coding for ubiquitously expressed protein Huntingtin (HTT), a large monomer of 350 kDa (Andrade and Bork, 1995).CAG expansion (above 37–40 repeats) in the exon1 of HTT results in the generation of an abnormally long polyglutamine tract (polyQ) in the N-terminus of the protein (The Huntington’s Disease Collaborative Research Group, 1993) that disturbs protein properties and makes it prone to form the insoluble aggregates, which are resistant to degradation by UPS and autophagy-related lysosomal proteolysis (Ding and Yin, 2008).A direct relationship between CAG repeat length and disease severity is observed in HD human (Paulson, 2018).Molecular events, such as protein aggregation, transcriptional dysregulation, and mitochondrial dysfunction, have been linked to HD pathogenesis (Kumar and Ratan, 2016).
In the HD brain, both major degradative proteostasis strategies UPS and autophagy are impaired.mHTT has been shown to inhibit proteasomal activity (Bence et al., 2001).Autophagy functions abnormally in HD as well,because mHTT prevents recognition and loading of cargo into developing autophagosomes.This results in the accumulation of “empty” autophagic vesicles (Martinez-Vicente et al., 2010).The failure of UPS and autophagy is followed by the accumulation of insoluble mHTT aggregates in HD central nervous system.
Visual deficit is one of complications of HD.HD patients manifest retinal thinning, temporal retinal nerve fiber layer thinning, loss of retinal ganglion cells, reduced visual evoked potentials, impaired color vision, and poor motion perception (Kersten et al., 2015; Andrade et al., 2016; Dhalla et al.,2019).Progressive retinal dysfunction and degeneration were also evidenced in animal models of HD (Helmlinger et al., 2002; Petrasch-Parwez et al., 2004;Batcha et al., 2012; Ragauskas et al., 2014; Dhalla et al., 2019).Among them,transgenic R6/1 mice, expressing human polyQ-expanded Htt exon 1 (115 CAG repeats) under human HTT promoter is the most popular reference model to study a mild late-onset HD (Mangiarini et al., 1996).R6/1 mice exhibit progressive retinal degeneration with an atypical pattern, wherein the destruction of outer and inner segments of photoreceptors does not result in photoreceptor cell death.Apoptosis is a rare event in the R6/1 retina(Helmlinger et al., 2002; Yefimova et al., 2021).
The ubiquitous pattern of HTT expression in all cells of the body determines the presence of normal HTT and its mutant counterpart in all neuronal and glial cells from the R6/1 retina (Helmlinger et al., 2002).Therefore, being exposed to proteotoxic stress induced by the expression of structurally damaged mHTT, each cell type from retinal population attempts to maintain its proteostasis.
The accumulation of aggregated forms of mHTT in all retinal layers (Helmlinger,2002) highlights the inability of retinal cells to manage abnormal mHTT.A notable exception is the retinal glial Müller cells, wherein no mHTT aggregates are detected bothin situ
andin vitro
(Helmlinger et al., 2002; Yefimova et al., 2021).To avoid the accumulation and further aggregation of toxic mHTT protein, Müller cells exert a particular non-degradative strategy to restore the dynamic equilibrium in the cytoplasm.This strategy relies upon myelinosome organelle, which loads mHTT on their membranes and exports the protein in the extracellular milieu, maintaining thereby their own proteostasis (Yefimova et al., 2021).In this regard, a high secretory activity of Müller cells, which play a critical role in the removal of metabolic “’wastes”’ (Reichenbach and Bringmann, 2013; Tworig and Feller, 2022) is their advantage over other retinal cells.“Switching on” of non-degradative MDS mechanism prevents the accumulation of mHTT-containing aggregates in secretory Müller cells in a situation where both classical strategies of degradative proteostasis (UPS and autophagy) failed (Figure 1).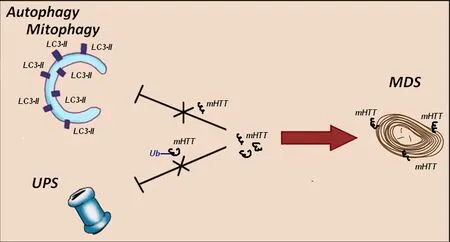
Figure 1 | In HD R6/1 retina myelinosome-driven secretion mechanism provides the export of mutant Huntingtin outside of retinal glial Müller cells.
In the R6/1 retina, myelinosomes are extremely frequent, indicating the importance of MDS mechanism for proteostasis maintenance in the HD retina.Myelinosomes are scattered throughout all retinal layers, being present not only in Müller glia but also in all types of retinal neurons containing mHTT inclusions and in intercellular spaces.Myelinosomes are encountered along the axons of photoreceptor cells and in the synaptic terminals, in the neurites of bipolar, horizontal, amacrine, and ganglion cells.In some cases,myelinosomes were found in close association with the membranes of ER corroborating previous observation by Prince et al.(1993).This suggests that ER membranes are a possible source for myelinosomes, generated/produced by retinal neurons (Figure 2).In our previous study, we demonstrated that myelinosomes were free of autophagy and exosome marker proteins(Yefimova et al., 2016).Furthermore, neuronal myelinosome pool can be replenished with the extrinsic myelinosomes, derived from Müller cells.Indeed, myelinosomes released in the extracellular milieu by Müller glia are extremely reactive organelles, which attack the neighboring cells, delivering them mHTT protein through membrane fusion (Yefimova et al., 2021).
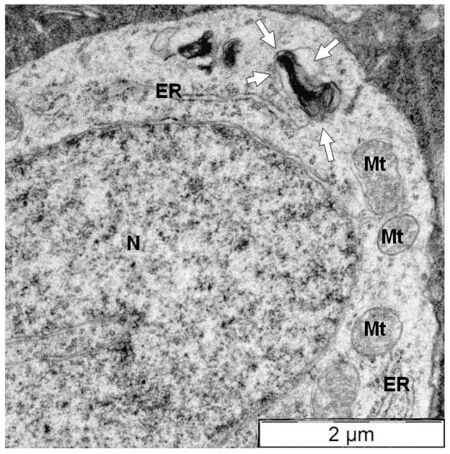
Figure 2| Direct connection between nascent myelinosome and endoplasmic reticulum in the cytoplasm of amacrine cell from the transgenic HD R6/1 mice retina.
Thus, it seems likely that in the R6/1 retina, only secretory Müller cells take advantage of MDS mechanism to maintain their proteostasis, whereas for retinal neurons with limited secretory activity, the MDS mechanism is ineffective.It should be noted, that the main roles of Müller cells are to support the neuronal function.These roles have been evolutionarily conserved across at least 300 million years, so that same general signature and function being observed from avians to reptiles and mammals (Pfeiffer et al., 2020).Any insufficiency of Müller cells can lead to dramatic consequences for retinal neurons, including retinal degeneration.Therefore, compared to retinal neurons, Müller cells are much more resistant to different insults as anoxia, hypoglycemia, or oxidative stress (Reichenbach and Bringmann,2013).In this line, the MDS mechanism seems to be once more evolutionary advantage of Müller cells, allowing them to maintain their proteostasis/health in order to support the viability and functional activity of retinal neurons.Nevertheless, being protective for Muller cells, the MDS mechanism can contribute to the spreading of mHTT over the HD retina due to the intercellular exchange of myelinosomes from glia-to-neurons.
Mitochondrial Damage, Oxidative Stress, and Myelinosomes in Huntington’s Disease Retina
A number of laboratories have provided evidence supporting the view on oxidative stress as one of key players in the pathogenesis of HD in central nervous system of human and animal models (reviewed by Kumar and Ratan, 2016).To our knowledge, no data on oxidative stress in HD retina are available in the literature.Mitochondria are a major source of ROS in mammalian cells.In non-pathologic situation, ROS are generated as normal byproducts of mitochondrial metabolism, resulting from the functioning of the electron transport chain for ATP production.Recent findings have placed ROS as signaling molecules involved in a variety of signal transduction pathways necessary for normal cellular functions (Kumar and Ratan, 2016).In a physiological situation, the level of ROS is strictly controlled by a special set of antioxidant enzymes and biomolecules.When a cellular antioxidant system becomes insufficient, a physiological level of ROS is overwhelmed,contributing to oxidative stress development.
Mitochondrial damage is one of the best-known causes of oxidative stress in various cell and tissue models.Mitochondrial dysfunction accompanies HD in human and animal models (Carmo et al., 2018).Mutant Htt localizes to the mitochondria, where it can either recruit soluble cytosolic proteins or interact with mitochondrial components (Guo et al., 2016).
HTT/mHTT is associated with mitochondria, being located on the cytosolic side of the mitochondrial outer membrane (Hamilton et al., 2020).The ultrastructural studies of cortical biopsies obtained from HD patients and HD rodent models showed abnormal mitochondria morphology (Carmo et al.,2018).Inefficient removal of damaged mitochondria is thought to contribute to the pathogenesis of HD and other neurodegenerative diseases (Šonský et al., 2021).
Striking morphological abnormalities of mitochondria, improper distribution of mitochondria in photoreceptor compartments, and the signs of mitochondrial degeneration are also a hallmark of HD R6/1 retina (Yefimova et al., 2021).Misplaced, abnormally swollen mitochondria were detected in perinuclear region of R6/1 photoreceptors.Degenerative mitochondria were frequent in RPE of HD mice.Therefore, an increase in ROS production in HD R6/1 retina seems plausible.As mentioned above, an oxidative stress triggers autophagy (Calandria et al., 2012), which plays a key role in elimination of damaged mitochondria.
Mitophagy is a selective autophagy responsible for quality control of mitochondria (Bakula and Scheibye-Knudsen, 2020).Mitophagy is impaired in HD through the direct interaction of mHTT with components of the mitophagic machinery (Guo et al., 2016).This leaves no appropriate operational mechanism responsible for the clearance of dysfunctional mitochondria (Šonský et al., 2021).Therefore, another mechanism is needed to efficiently remove damaged mitochondria from HD retina.Our data show that myelinosomes can contribute to elimination of damaged mitochondria as well.TEM examination of R6/1 retina evidenced the particular mode of mitochondrial destruction in RPE cells, wherein mitochondrial membranes were thoroughly debobinated andde novo
re-bobinated forming myelinosome-like structure (Figure 3).Of note, a study from an animal model of experimental autoimmune encephalomyelitis defined myelinosomes as local myelin out-foldings, arising at the early stage of myelin damage and dissociated from myelinated axons(Romanelly et al., 2016).Thus, the data presented suggest that myelinosomes are also involved in catabolic processes related to “dismantling/disassembly”of damaged sub-cellular structures (mitochondria, myelin sheath), which occurs independently of lysosomal proteolysis (Figure 4).Thus, ubiquitous expression of mHTT/HTT in all cell types of the retina determines the fundamental importance of the HD R6/1 model, wherein each cell presents its proteostasis strategies to cope with aberrant proteins and damaged organelles.
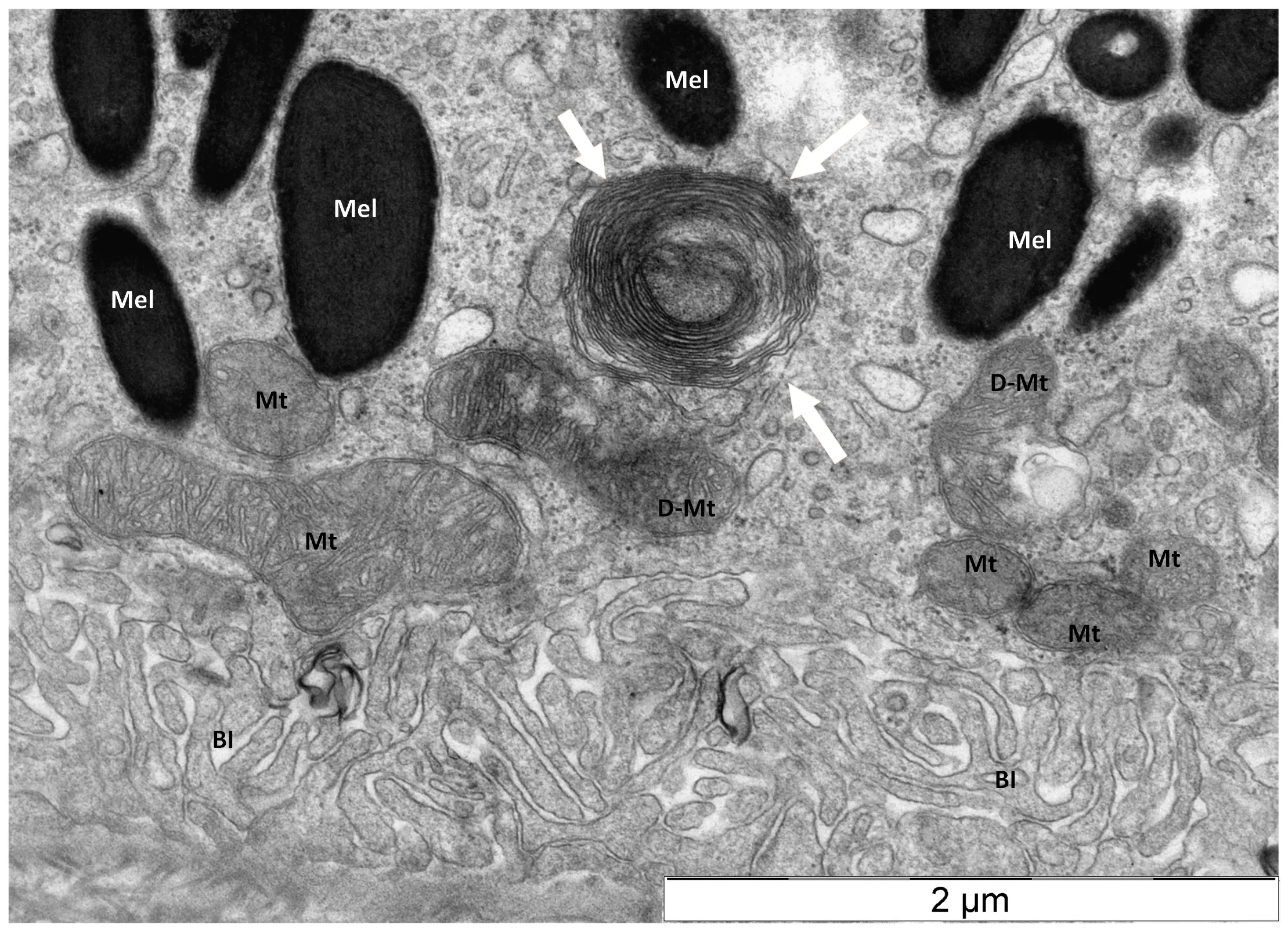
Figure 3 | Destruction of damaged mitochondrion in retinal pigmented epithelium of transgenic HD R6/1 model mice.
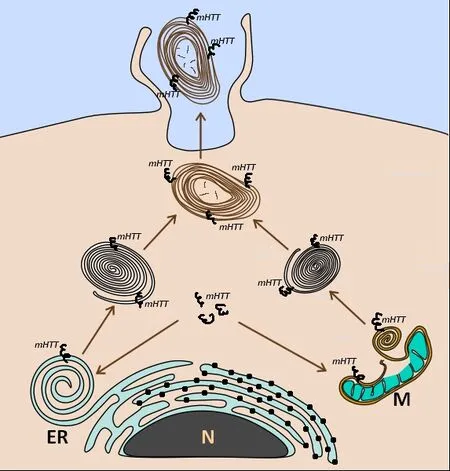
Figure 4 | Possible sources of myelinosomes in ocular tissues of HD R6/1 mice.
Royal College of Surgeons Rats
In contrast to R6/1 mice, expressing a ubiquitous protein HTT in all cell types of the retina, Royal College of Surgeons (RCS) rat is a striking example of how a single protein mutation in a single cell type can cause a detrimental effect on multiple cellular processes dependent on proteostasis both inside and outside the cell (Paraoan et al., 2020).
RCS rat is an animal model of retinitis pigmentosa (RP), a group of rare eye diseases that affects the retina, causing vision loss through progressive degeneration of retinal neurons.RCS rats are a naturally occurring rat strain that was described in the first half of the 20century (Bourne et al., 1938).RCS rats bear mutation in the Mertk receptor tyrosine kinase (Nandrot et al., 2000).The deletion in the Mertk cDNA, resulting from a large genomic deletion (D’Cruz et al., 2000) led to the production of non-functional 20-amino-acid Mertk proteins in RPE cells.MERTK, a member of the TAM family of receptor tyrosine kinases, plays a pivotal role in the elimination of tips of photoreceptor outer segments (POS) through unconventional autophagy-assisted phagocytosis process (LC3-associated phagocytosis)(Almedawar et al., 2020).The inability of RPE cells to ingest the tips of POS results in irreversible blindness.The pattern of degeneration in RCS rats has been thoroughly documented using histological and electron-microscopy techniques.A progressive development of debris zone of uningested POS in the subretinal space of RCS retina by 3 weeks of age is followed by complete loss of the outer segment area and further apoptotic death of photoreceptors.Secondary to photoreceptor degeneration is the neurodegenerative changes in the retina including the loss of retinal ganglion cells, withdrawal of bipolar cell axons, Müller cell gliosis, neuronal sprouting of horizontal cells, and microglial activation (reviewed by Vollrath et al., 2001).
One of the earliest events during retinal degeneration in RCS rats is the formation of “lamellar whorls” which are the electron-dense lamellar membranes located in the subretinal space of RCS retina.An elegant study by LaVail et al.(1972) using TEM radioautography showed that the RPE cells produce an extra lamellar material.2 hours post-injection of amino-acids-H3,newly synthesized proteins were displaced from cell soma to lamellar whorls/myelinosomes located in the subretinal space.Extracellular lamellar whorls/myelinosomes were also detected in the subretinal space of Mertk KO mice,exhibiting an RCS-like phenotype (Duncan et al., 2003).In both studies, the lamellar whorls/myelinosomes were produced by living cells and were not considered as “dead” material (LaVail et al., 1972).
Intracellular lamellar whorls/myelinosomes were also a hallmark of RPE cytoplasm in RCS, but not in control rats.In some preparations, the association of lamellar whorls/myelinosomes with the membranes of ER was observed likewise, as in HD R6/1 mice (Figure 2).Sometimes the lamellarwhorls/myelinosomes were detected between the adjacent RPE cells,indicating that lateral directions can also be used to address the organelles outwards of the cells.Nevertheless, myelinosomes did not seem to cross the basal lamina (LaVail et al., 1972).
In contrast to ubiquitous protein HTT, MERTK is expressed by RPE cells only.Therefore, it remains to be determined whether lamellar whorls/myelinosomes from RCS RPE carry non-functional 20-amino-acid Mertk proteins.Some interesting information on myelinosome content was acquired from the study of iron metabolism in RCS retina.Proton-induced X-ray emission spectra analysis and TEM analysis for the presence of iron revealed large deposits of toxic unbound iron in lamellar whorls/myelinosomes in subretinal space of RCS rat retinas (Sergeant et al., 2001).Unbound iron facing photoreceptor tips and mitochondrial dysfunction (Zhu et al., 2017) are likely the main triggers of oxidative stress in RCS rat retina, which contributes to the apoptosis of photoreceptors (Camaschella et al., 2020).
It should be noted that according to the study by LaVail et al.(1972),degenerative photoreceptors from RCS retina can also contribute to subretinal pool of myelinosomes.In this case, we cannot exclude the oxidative stress/oxidative damage as a source of aberrant protein production in photoreceptor cells of RCS rats (see section on Light-Induced Retinal Degeneration).
Retinitis Pigmentosa Caused by Rhodopsin Mutations
Rhodopsin is the light receptor and a major constituent of the rod outer segment of photoreceptor cells.It is a member of the G protein-coupled receptor family of cell surface proteins, which consists of the apoprotein opsin covalently bound to the chromophore 11-cis retinal.Light causes the isomerization of 11-cis retinal to all-trans retinal, which activates rhodopsin and initiates the G protein-mediated signaling cascade called phototransduction (Lenahan et al., 2020).
Over 100 mutations in rhodopsin have been determined to cause inherited retinal disease, most causing autosomal dominant RP.A number of these mutations result in protein misfolding and aggregation (reviewed by Athanasiou et al., 2018).The first discovered mutation in rhodopsin was the P23H mutation (Dryja et al., 1990).Being the most common mutation in the United States, this mutation results in the substitution of histidine for proline at amino acid residue 23 (RHOP23H) (Sohocki et al., 2001).It has been demonstrated that the P23H variant of rhodopsin activates ER stress and unfolded protein response, leading to activation of both the proteasome and autophagy pathways (reviewed by Athanasiou et al., 2018).P23H transgenic rodent models were designed to mimic autosomal dominant RP, displaying compromised rhodopsin trafficking through the endoplasmic reticulum to the outer segment.The photoreceptors die through a non-apoptotic mechanism secondary to the oxidative stress caused by the rhodopsin mutation (reviewed by Athanasiou et al., 2018).Exploring the fate of misfolded rhodopsin using cell models (Saliba et al., 2002) as well as homozygous RHOP23H transgenic mice showed that both autophagy and proteasome are implicated in the management of aberrant proteins (Yao et al., 2018; Sen et al., 2021).However, experimental data on the effect of these degradation pathways on preservation of P23H retina are conflicting.Thus, the study by Yao et al.(2018)showed that autophagy contributes to photoreceptor cell death in P23H mice,and that decreasing autophagy shifts the degradation of misfolded rhodopsin protein to the proteasome is protective.On the contrary, a study by Sen et al.(2021) found that the inhibition of proteasome favors cell survival and suppress P23H retinal degeneration in P23H rat retinal explants.
In the context of this review, it is noteworthy that unusual electron-dense membranous “disk-like structures”/(myelinosomes) were detected in photoreceptor inner segments of P23H mice (Sakami et al., 2014), suggesting the involvement of myelinosomes in proteostasis maintenance in this model.The presence of myelinosome-like membranous structures was also reported in other rodent models carrying mutation in rhodopsin gene as Q344ter mutation (Concepcion and Chen, 2010) and Ter349Glu mutation(Hollingsworth and Gross, 2013).
Histological examination of retinal sections from human eyes with rhodopsin Glu181Lys mutation, and with Arg2310Gly mutation showed a loss of rod photoreceptors.Remaining cone photoreceptors contained the intracellular perinuclear “membraneous swirls” revealed by TEM analysis (To et al., 2004).An extracellular myelinosome called “unidentified membrane structure”was also detected in the interphotoreceptor matrix from retinal section of a patient carrying Q64ter Rhodopsin mutation (Milam et al., 1996).Collectively,retinal degeneration caused by different mutations in rhodopsin gene in human and animal models of RP is associated with myelinosome production,detected in both intra- and extra-cellular compartments.Further studies are needed to understand whether retinal cells other than rod and cone photoreceptors contribute to myelinosome production in degenerative retinas with mutations of rhodopsin gene and what are the cargoes carried by myelinosomes.
Light-Induced Retinal Degeneration
By its action on rhodopsin, light triggers the visual transduction cascade but can also induce cell damage and death through phototoxic mechanisms.During acute light exposure, excessive generation of ROS by excessive action of visual cycle cannot be regulated by the self-defense system and results in photoreceptor cell death (Ozawa, 2020).
Histological examination immediately after light exposure reveals that photoreceptor cell damage is initiated in the distal tips of POS and that with time it progresses to include the entire POS.The damage of RPE evolves secondary to POS damage.A unifying factor in photoreceptor/RPE damage appears to be light-induced reactive oxygen species, generated by the bleaching of rhodopsin or from compounds in the RPE, such as bisretinaldehyde-phosphatidylethanolamine (A2E) (Ozawa, 2020).Enhanced autophagic events have been documented under light exposure in RPE and photoreceptor cells bothin vitro
andin vivo
techniques, suggesting a role for autophagy in RPE and neural retina proteostasis (Chen et al., 2016).Nevertheless, to date, it remains unclear whether autophagy is involved in cell protection or the induction of autophagic cell death (Chen et al., 2013;Chen et al., 2016).The inner retina seems to be rather resistant to light-induced damage while Müller cell gliosis is noticed by numerous investigators (Reichenbach and Bringmann, 2013; Riccitelli et al., 2021).Studies from albino light damage model showed that photoreceptor cell death was not associated with an increase in global protein ubiquitination or unfolded protein response,suggesting that proteasome degradation does not play a primordial role in proteostasis maintenance in this model (Wang and Chen, 2014).UPS degradation is of extreme importance for proteostasis maintenance in the inner retina, which is less vulnerable to light-induced damage (Kageyama et al., 2019).
Because the outer retina is more vulnerable to light exposure than the inner retina, the emergence of myelinosomes in RPE-POS area seems plausible.Indeed, TEM examination of retinal sections from albino rats exposed to bright light pointed out the presence of “lamellar inclusion bodies” in RPE and in interphotoreceptor matrix in close proximity of RPE cells (Kuwabara, 1970).In vitro
study of primary RPE cells irradiated with blue light revealed “large whorls of membrane or whorled inclusions” (Pang et al., 1999).Remarkably,the authors noticed that some whorls could result from dismantlement of damaged mitochondria, likewise what we detected in HD R6/1 retina (Figure 3).Electron-dense “membranous structures” were also discovered in the neuroretina of mice exposed to bright light irradiation (Chen et al., 2013)as well as in light-irradiated Drosophila retinas, where they were termed electron-dense “multilamelate bodies” (Stark and Carlson, 1984).Thus,myelinosome production is a characteristic of light-induced retinal damage in different species.Stargardt Disease, Spinocerebellar Ataxia7,Mutation in PRPC8 Gene
Analysis of literature data allows concluding that myelinosome production is associated with various degenerative states of the retina.Thus, myelinosomes called “membrane swirls” were found in cone photoreceptors of a rodent model of RP caused by the mutation inPRPC8
gene (encoding the pre-mRNA processing factor 8 that is the homolog of yeast Prp8) (To et al., 2004).Myelinosomes termed as “vesiculated membranes” were also detected in the retina of spinocerebellar ataxia type 7 (SCA7) mice (Niewiadomska-Cimicka et al, 2021).Alike HD, SCA7 belongs to the class of repeat expansion diseases.In SCA7 mice, the CAG expansion affects the gene of ataxin-7, coding for the ataxin 7 protein, a component of the STAGA transcription coactivator complex.The SCA7 R7E transgenic mouse expresses the human ataxin-7 with a polyQ expansion (90Q) under rhodopsin promoter control and thereby targets the expression in rod photoreceptors only (Yvert et al., 2000).In SCA7 mice “vesiculated membranes”/myelinosomes were found in the subretinal space, suggesting their emission from photoreceptors (Niewiadomska-Cimicka et al, 2021).Of note is that the SCA7 retinal degeneration exhibits an atypical pattern closely resembling those in HD R6/1 (Helmlinger et al., 2002;Yefimova et al., 2021).This highlights a key role of CAG expansion in atypical pattern of retinal degeneration, suggesting a polyQ-expanded ataxin7 as a potential cargo of myelinosomes in SCA7 retina.
Stargardt disease is one of the most frequent inherited macular dystrophies in humans caused by mutations in the ATP-binding cassette A4 (ABCA4)gene.TheABCA4
gene encodes for a transmembrane protein located in the rim of photoreceptor disks (Allikmets et al., 1997) and in RPE cells (Lenis et al., 2018).ABCA4 is involved in all-trans retinal transports through the photoreceptor disk membrane and is thus part of the visual cycle (Taubitz et al., 2018).TEM sections from the outer retina of rodent models of Stargardt disease show multiple myelinosome-like structures in photoreceptors and RPE cells, as well as in the subretinal space (Taubitz et al., 2018), suggesting the involvement of myelinosome-based mechanism for proteostasis maintenance in this disorder.Retinal Degenerations Induced by the Ablation of Autophagy Genes
Accumulation of myelinosome-like structures is a hallmark of retinal degeneration phenotypes resulting from ablations of autophagy and/or proteasome genes.This is an expected phenomenon, aimed at maintaining proteostasis by a non-degradative strategy in a situation where degradative strategies have failed.Thus, suppression of autophagy activation specifically within RPE through the deletion of autophagy inducer RB1CC1 results inthe degeneration of the retinal pigmented epithelium.Degenerative RPE exhibits vacuolation and accumulation of myelinosome-like structures called “unprocessed phagosomes” which rather resembled debobinated mitochondria from Figure 3, and the subretinal drusenoid deposits (Yao et al.,2015).
RPE-specific deletion of autophagy genesAtg5
orAtg7
in mice causes an ageprogressive RPE phenotype, with electron-dense myelinosome-like structures in RPE cytoplasm and “cytosolic debris” (Perusek et al., 2015; Zhang et al.,2017).Deletion of essential autophagy genes, includingBeclin1
systemically orAtg7
in only rod photoreceptors, resulted in increased susceptibility to light-induced retinal damage and formation of myelinosome-like structures(Chen et al., 2013).Deletion of mitophagy essential genePark
also resulted in increased susceptibility of RPE to light.Ultra-structural analyses showed mitochondrial and endoplasmic reticulum impairment after light exposure and presence of whorled membranes in cytoplasm of RPE cells (Chen et al.,2013).Retinal proteasome activities are known to decline with age, and impaired proteasome function was reported to induce photoreceptor degeneration(reviewed by Gunawan et al., 2021).According to our knowledge, the timecourse of retinal degenerations induced by proteasome impairment is explored by light-microscopy only.Therefore, TEM studies are needed to clarify the involvement of myelinosome-based strategy in retinal disorders caused by proteasome insufficiency.
Age-Related Macular Degeneration
ARMD is a leading cause of blindness in the western world (Wang et al., 2009; Golestaneh et al., 2017).Ample recent evidence implicates dysregulated proteostasis as a key factor in the development of ARMD.ARMD is classified as either dry (atrophic), or wet (exudative) form.The formation of drusen (yellow aggregates in the macula between the RPE and Bruch’s membrane) are hallmarks of both ARMD phenotypes, dry and wet.Choroidal neovascularization is the hallmark of wet ARMD (Paraoan et al., 2020).ARMD is a complex eye disease with many pathogenesis factors,including defective cellular waste management in RPE.RPE dysfunction is a prominent event in the pathogenesis of both ARMD forms, so that the issue on proteostasis maintenance by RPE cells is a promising area of research.As widely accepted, waste components as unfolded, damaged and unneeded proteins are degraded and recycled in RPE cells by two main machineries—UPS and autophagy (Blasiak et al., 2019).A third non-degradative strategy of waste proteins cleaning was reportedin vitro
using aged RPE model (Wang et al., 2009).In this model, an increased mitochondrial damage was induced by rotenone, an inhibitor of mitochondrial complex I.The damage was followed by a decrease of lysosomal activity and a concomitant increase of exosomal release (Wang et al., 2009).This highlights the importance of non-degradative strategy of proteastasis maintenance when degradative pathway fails due to lysosomal dysfunction.Some evidence suggests a possible contribution of myelinosomes to drusen formation.ARMD-like phenotype resembling a wet form of disease is exhibited by Cu, Zn-superoxide-dismutase (SOD1) null mice (Imamura et al.,2006).Cu, Zn-SOD1 is one of the main antioxidant enzymes in the retina.TEM examination of drusen from RPE/choroid preparations of SOD1 null mice revealed electron-dense myelinosome-like membranes termed “debris-like”membranes (Imamura et al., 2006).“Membranous profiles” with electrondense exteriors and moderately electron-dense interiors are described in postmortem human eye from ARMD human (Curcio, 2018).According to authors, these “membranous profiles” can represent partly preserved lipoprotein particles.The size of these particles is around 200-300 nm,which corresponds to an average size of myelinosomes.Myelinosome-like structures were also described in drusen formed in patient-derived hiPSCRPE models of macular dystrophy (Galloway et al., 2017; https://www.urmc.rochester.edu/eye-institute/research/labs/singh/projects/extracellular-matrixalteration.aspx).The authors of this study concluded that the dysfunction of RPE cells alone is sufficient for the initiation of sub-RPE drusen formation in these diseases.Altogether, the contribution of myelinosomes in drusen formation cannot be ruled out.
Alzheimer’s Retinopathy and Glaucoma:Degenerative Changes in the Inner Retina
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder affecting over 50 million people worldwide.A cognitive decline including disorientation, short-term memory loss, confusion, and sociobehavioral impairments are the hallmarks of AD.Basal forebrain cholinergic neurons are the most damaged in the AD brain (Mirzaei et al., 2020).The pathogenic mechanisms of AD remain unclear.The disease is associated with brain proteostasis disorders manifested by the accumulation of insoluble protein aggregates: the intracellular tangles of phosphorylated tau protein(p-tau) and extracellular plaques of amyloid Aβ peptide – a derivative from the cleavage of amyloid precursor protein (APP) (Hou et al., 2020).Both amyloid Aβ and tau are intrinsically disordered proteins, which lack a definite three-dimensional structure (Subedi et al., 2022).Oxidative stress,mitochondrial dysfunctions, deregulations of UPS, and autophagy in the AD brain are noticed by numerous investigators (Zare-Shahabadi et al., 2015;Hegde et al., 2019; Hou et al., 2020).
Studies of fine structure of amyloid plaques in AD brain by TEM recognized to contain extracellular aggregates of the amyloid Aβ peptide surrounded by microglia and swollen axons.The latter are filled with membranous electron-dense structures/myelinosomes, resembling “lysosome organelles or their hybrids” (Dikranian, 2012; Gowrishankar et al., 2017).These data corroborated the study by Hartmann et al.(1997) on the distinct sites of the production of Aβ peptide in the neuronal and non-neuronal cells.According to authors, the non-neuronal cells (fibroblasts) produced significant amounts of Aβ only at cell surface, whereas in neuronal cells Aβ generation occurs in the cytoplasm.Immuno-electron microscopy revealed the endoplasmic reticulum,the trans-Golgi network and the multilamellar myelinosome-like vesicles as Aβ-harboring structures in hippocampal neurons (Hartmann et al., 1997).Thus, a part of intraneuronal Aβ is bound to myelinosomes/myelinosomelike vesicles.Taking into consideration the involvement of myelinosomes in mHTT traffic along the neuronal cells (Yefimova et al., 2021), a transneuronal propagation of myelinosome-bound Aβ seems likely.The electron-dense myelinosome-like structures can be also discerned in intracellular tangles of neurons from transgenic mice expressing human P301S tau protein (Allen et al., 2002).
AD affects a neurosensory retina, so that the deposition of protein aggregates in the retina is detected prior to brain (Mirzaei et al., 2020).In contrast to degenerative states of the retina described above, AD mostly targets the inner retina.Degeneration of GCL and thinning of the nerve fiber layer are the hallmarks of AD retinas from patients and animal models (Mirzaei et al.,2020).Retinal pathology in AD closely resembles those in glaucoma - a major cause of blindness worldwide, linked to raised intraocular pressure.Like AD retinopathy, glaucoma affects the inner retina and is characterized by the progressive degeneration of retinal ganglion cells (RGC) and the optic nerve,so that some investigators have proposed that glaucoma is an ocular AD(McKinnon, 2003).
The presence of Aβ plaques and neurofibrillary tangles was reported for both AD and glaucomatous retinas (Gupta et al., 2008; Yan et al., 2017; den Haan et al., 2018).Nevertheless, a careful histological study of post-mortem AD retinas revealed no typical Aβ plaques and neurofibrillary tangles like in the cerebral cortex (den Haan et al., 2018).This indicates that retinal manifestations of AD pathology are different compared to cerebral manifestations (den Haan et al., 2018).Taking into account that the aggregation state of Aβ plays an important role in Aβ-induced pathology (Dahlgren et al., 2002), a thorough examination of AD retinas by TEM and immuno-electron microscopy is needed to understand to which extent the MDS mechanism can be involved in the pathological process, and whether the myelinosome’s role concerns the traffic/evacuation of intrinsically disordered Aβ peptide and/or tau protein, or other aberrant proteins produced under oxidative stress/oxidative conditions in AD and glaucoma (Kimura et al., 2017; Cassidy et al., 2020).Respectively,the implication of myelinosomes in the destruction of damaged mitochondria(Hou et al., 2020) and myelinated axons from the myelinated region of optic nerve (Hinton et al., 1986) cannot be either ruled out.
Along with Aβ plaques, the typical sub-RPE drusen (Ukalovic et al., 2018)and atypical amyloid deposits above the RPE (Koronyo et al., 2017) were detected in AD individuals by different techniques.Because the sub-RPE drusen is known to contain the amyloid Aβ protein (Anderson et al., 2004),the AD animal models are used to study the pathogenesis of both AD and ARMD.5XFAD mice, expressing human APP and preselenin 1 transgenes with a total of five AD-linked mutations and used to study the pathogenesis of AD,develop an Aβ-containing sub-RPE drusen (Park et al., 2017).Remarkably,the cytoplasm of RPE cells of 5XFAD mice is filled with cystic vacuoles,holding a myelinosome-like membranous material.According to authors,a rupture of such vacuoles results in the dissemination of membranous material throughout the RPE (Park et al., 2017).Thus, at least for RPE cells,the implication of myelinosomes in the pathological process in the AD retina seems likely.
In glaucoma patients, an optic disc drusen is often detected (Gramer et al.,2017).It localizes within the optic nerve head, formed by unmyelinated segments of RGC axons (Zhu et al., 2018).Both APP and amyloid Aβ peptide localize in RGC, and the levels of Aβ are increased in glaucomatous RGC compared to control.Moreover, in experimental models, Aβ induces RGC apoptosis, while targeting different components of Aβ formation and aggregation pathway reduces RGC apoptosisin vivo
(Guo et al., 2007).As believed, the unmyelinated segment of RGC axons is the locus of initial insult to RGC axons in glaucoma (Zhu et al., 2018).TEM examination of this segment from glaucomatous mice optic nerve head revealed the electrondense membranous whorls (Lampert and Vogel, 1968) or multilamellar structures/myelinosomes (Zhu et al., 2018) located in the vicinity of damaged mitochondria.This is in agreement with recent evidence showing a mitophagy deficit in murine glaucomatous optic nerve (Duarte, 2021).Therefore, the contribution of myelinosomes to mitochondrial dismantling as occurs in R6/1 RPE (Figure 3) seems likely.Myelinosome-like membranes were also abundant in corpora amylacea of optic nerve head of glaucomatous eyes from affected patients (Tso, 1981).Interestingly, tau is among the main proteins of corpora amylacea which are present in glaucomatous and AD retinas as well (den Haan et al., 2018).In normal retina, tau protein is mainly detected in plexiform layers and in the RPE (den Haan et al., 2018; Mirzaei et al., 2020).An abnormal p-tau locates in the horizontal cells of glaucomatous retinas (Gupta et al., 2008).Interestingly, the levels of tau dramatically increased in the vitreous body of glaucoma patients (Yoneda et al., 2005), while the levels of Aβ decreased.The decrease of vitreal Aβ levels is thought to be related to Aβ deposition in theglaucomatous retina.Such as interpretation cannot be applied for tau protein,suggesting that different pathways operate in the eye to evacuate different abnormal proteins generated under pathological conditions.To which extent myelinosomes contribute to these pathways remains to be determined.
The Traffic of Myelinosomes in the Retina
As already noticed, in degenerative HD R6/1 retina, myelinosomes were produced by all types of retinal neurons and secreted outwards by Müller glia, which is known to redistribute metabolic waste into the blood and the vitreous body (Reichenbach and Bringmann, 2013).Thus, in a situation where recycling of the aberrant protein is not possible due to the failure of degradation strategies, the non-degrading myelinosome-based mechanisms reject the toxic mHTT protein out of the cells.This suggests that special evacuation routes operate in the eye, which is devoid of traditional lymphatic drainage.
A recent study on intraocular traffic in rodents using HiLyte-594–tagged human Aβ tracer uncovered a new glymphatic/lymphatic route for fluid and waste clearance, which provides the cleaning of Aβ from the inner retina(Wang et al., 2020).As known, the intravitreal-injected tracers were found in the cervical lymph nodes (Wang et al., 2020).Wang et al.(2020) followed Aβ tracer after intravitreal administration.The data obtained indicated that Aβ tracer exited the eye along the optic nerve.The transport of Aβ was restricted to RGC axons, consistent with the avid uptake of Aβ by neurons (Kanekiyo et al., 2013).After having crossed the lamina cribrosa barrier, an intra-axonal Aβ was cleared via the perivenous space and subsequently drained to lymphatic vessels (Wang et al., 2020).Of note is that the clearance of Aβ was dependent on glial water channel aquaporin-4 (AQP4) expressed by Müller cells as shown usingAqp4
andAqp4
mice.In rodent models of glaucoma Aβ leaked from the eye via defects in lamina barrier instead of directional axonal efflux.Therefore, the authors suggest that the abnormal transport of APP/Aβ or other metabolites contributes to degeneration of RGC axons in glaucoma(Wang et al., 2020).In the context of this review, it should be noted that the temporal retinal nerve fiber layer thinning and the loss of RGC were also revealed in HD patients(Kersten et al., 2015; Andrade et al., 2016; Dhalla et al., 2019), suggesting the similar scenario of RGC loss induced by abnormal transport of toxic mHTT protein.Remarkably, during the preparation of this review, a new datum on ultrastructure of HD R6/2 optic nerve has been published (Mazur-Michałek et al., 2022).Compared to R6/1 mice, R6/2 mice manifest the accelerated form of the disease and a more severe retinal phenotype (Helmlinger et al., 2002).TEM examination of the optic nerve revealed the thinning of myelin sheath and the presence of myelinosomes called “myelonoid bodies”, supporting thereby the idea on the involvement of myelinosomes in pathological process in RGC axons, which form the optic nerve.Again, one cannot exclude the presence of myelinosomes in the vitreous body of R6/1 retina.As usual,eye dissection protocol for EM examination of the retina requires complete removal of the vitreous body.Thus, other protocols are needed for further analysis of vitreous body for myelinosome presence.
In all pathological states of the outer retina described above, myelinosomes were encountered in the subretinal space.This undoubtedly disturbs proteostasis in the interphotoreceptor matrix (Sørensen, 2019).As for R6/1 retina, it seems likely that both Müller cells and secretory RPE cells contribute to the subretinal myelinosome pool (Figure 5).
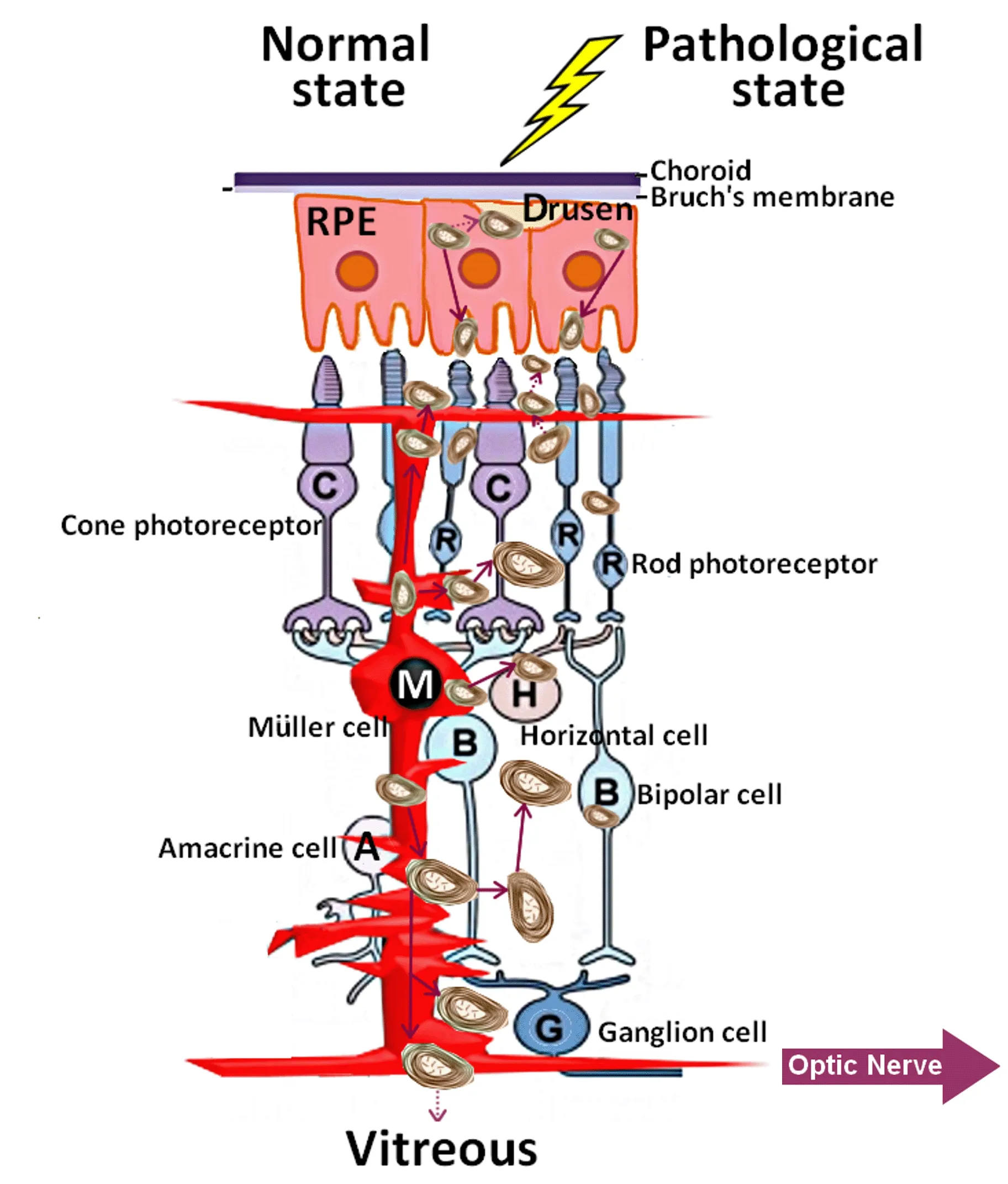
Figure 5|Myelinosome traffic in the pathological retinas.
For other degenerative states of the outer retina, the arguments are in favor of RPE cells and probably for photoreceptors (Milam et al., 1996;LaVail et al., 1972).As widely accepted, the substantial metabolic waste generated in the outer retina is removed via the underlying choriocapillaris(Keeling et al., 2018).Nevertheless, in R6/1 retina, no myelinosomes were detected in this compartment.Studies from other retinal pathologies indicated that myelinosome-like structures accumulate in sub-RPE-basal lamina space (Curcio, 2018).In this line, exploring of other ocular fluids(aqueous humor, tears) as well as cervical lymph nodes for the presence of myelinosomes should clarify the possible roads of their evacuation from the eye.Understanding of evacuation pathways is an important issue,because myelinosome organelles loaded with aberrant proteins, move in retinal fluids, exchange between different cell populations, and contribute thereby to disease spreading over the retina.In this context, it is worth to note that myelinosomes loaded with the mHTT protein were also abundant in pathological HD R6/1 testis (Yefimova et al., 2016).Myelinosome-like organelles were recently found in human seminal plasma, suggesting that evacuation of myelinosomes does proceed through biological fluids (Yefimova et al., 2020).
Conclusion
Timely and efficient elimination of aberrant proteins and damaged organelles,formed in response to various genetic and environmental stressors, is a vital need for all cells of the body.In ocular tissues, both degradative and nondegradative strategies are combined to maintain proteostasis.When recycling of aberrant proteins and damaged organelles (mitochondria) is not possible due to the failure of degradation strategies, the non-degradative MDS mechanism contributes to proteostasis maintenance in the neuroretina and RPE.The presence of myelinosomes/myelinosome-like organelles in various pathological states of the retina in different species (flies-rodents-humans)highlights the importance of MDS mechanism for ocular pathophysiology.Being protective for secretory Müller cells and RPE cells, MDS mechanism seems to be less effective for retinal neurons with limited secretory activity.Furthermore, intercellular exchange of myelinosomes can engage the spreading of metabolic “waste” over the retina and sub-RPE-basal lamina space.Exploring the fundamental aspects of myelinosome biogenesis,characterization of the molecular fingerprints of these organelles and the molecular mechanism of myelinosome-mediated sorting and transfer of cargo proteins will contribute to providing important basic resources to counteract several vision pathologies.
Acknowledgments:
I thank Dr.J.C.Hervé (Inserm UMR-1082 Irtomit, Poitiers,France), Dr.K.Mérienne (Laboratoire LNCA, CNRS, Université de Strasbourg,France) and Prof.N.Bourmeyster (Laboratoire STIM CNRS ERL 7003,Université de Poitiers, France) for critical reading of the manuscript.I thank Dr.E.Béré (Plateforme IMAGE-UP, Université de Poitiers, France) and Image UP platform from Poitiers University for electron microscopy experiments.I thank Ms A.Yefimova (State University of Industrial Technologies and Design, Saint-Petersburg, Russia) for her help in manuscript illustration.
Author contributions:
MGY designed the concept of the manuscript,contributed to literature collection and data analysis, wrote and edited the manuscript, and approved the final version of the manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
- 中国神经再生研究(英文版)的其它文章
- Patient-specific monocyte-derived microglia as a screening tool for neurodegenerative diseases
- Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases
- Inflammation in diabetic retinopathy: possible roles in pathogenesis and potential implications for therapy
- Targeting the nitric oxide/cGMP signaling pathway to treat chronic pain
- Neurosteroids as stress modulators and neurotherapeutics: lessons from the retina
- Anti-IgLON5 disease: a novel topic beyond neuroimmunology