
Endocannabinoid control of neuroinflammation in traumatic brain injury by monoacylglycerol lipase in astrocytes
2022-09-27 09:44ChuChen
中国神经再生研究(英文版) 2023年5期
Chu Chen
Traumatic brain injury (TBI) is a temporary or permanent disruption of brain function caused by external forces.TBI has been recognized as an important risk factor for the development of Alzheimer’s disease and dementia later in life.However, the mechanisms by which TBI contributes to developing Alzheimer’s disease are largely unknown.In particular, no effective therapies are currently available for the prevention and treatment of TBI-induced neurodegenerative disease.The acute brain damage after TBI results not only from primary injury, which is the result of the external mechanical force but also from secondary injury, which is associated with a complex cascade of molecular, cellular, and immune responses.Neuroinflammation associated with other processes plays a critical role in causing secondary injury following TBI (Simon et al., 2017).The extent of neuroinflammatory responses seems to be closely correlated with the outcome following TBI (Woodcock and Morganti-Kossmann,2013).This means that while the primary injury immediately following TBI is not preventable,appropriate and timely intervention to resolve neuroinflammation following the primary injury would be the key to preventing further brain damage, neuropathological changes, and synaptic and cognitive impairments (Zhang et al., 2015).
Endocannabinoids are endogenous lipid signaling mediators involved in a variety of physiological,pharmacological, and pathological processes.2-Arachidonoylglycerol (2-AG), the most abundant endocannabinoid and full agonist for cannabinoid (CB) 1 and CB2 receptors, displays anti-inflammatory and neuroprotective properties in response to proinflammatory, neurotoxic, and mechanical insults (Panikashvili et al., 2001; Du et al., 2011; Nomura et al., 2011; Chen et al.,2012; Zhang et al., 2014, 2015; Hu et al., 2022).Earlier studies provided evidence that 2-AG is capable of protecting the brain from a closed head injury (Panikashvili et al., 2001), which is largely associated with the suppression of neuroinflammatory responses (Panikashvili et al.,2005; Zhang et al., 2015), suggesting that 2-AG is an endogenous terminator of inflammation in response to harmful insults that elicit inflammatory responses.However, 2-AG is rapidly hydrolyzed by several enzymes, including monoacylglycerol lipase (MAGL), α/β hydrolase domain-containing protein 6 and 12, and cyclooxygenase-2.In the brain, 2-AG is predominantly degraded by MAGL(Blankman et al., 2007).The immediate metabolite of 2-AG is arachidonic acid, which is a precursor of prostaglandins and leukotrienes.Certain types of prostaglandins (i.e., prostaglandin E2) and leukotrienes are proinflammatory mediators and neurotoxic.Thus, “a dual hit” occurs when MAGL is inactivated, resulting in augmentation of anti-inflammatory and neuroprotective 2-AG signaling while lowering pro-inflammatory and neurotoxic eicosanoids (Hu et al., 2022).Indeed,the results from previous studies indicate that pharmacological inactivation of MAGL resolves neuroinflammation and attenuates neuropathology as well as improves synaptic and cognitive functions in several animal models for brain disorders, including Alzheimer’s disease and TBI (Chen et al., 2012; Piro et al., 2012, 2018;Zhang et al., 2014, 2015; Mayeux et al., 2017;Hashem et al., 2021).These neuroprotective effects produced by restraining 2-AG degradation result likely from enhanced 2-AG signaling and concurrently decreased eicosanoid levels (Nomura et al., 2011), suggesting that MAGL plays an important role in maintaining brain homeostasis and terminating neuroinflammation that occurs in neurodegenerative diseases by regulation of 2-AG metabolism.
Although inhibition of 2-AG metabolism by pharmacological inactivation of MAGL resolves neuroinflammation, reduces neuropathology, and improves synaptic and cognitive functions in TBI,the molecular mechanisms by which inactivation of MAGL exerts neuroprotective effects against brain trauma remain unclear.The latest study by Hu et al.(2022) shed new light on this unsolved issue.By using amgll
knockout mouse model, the authors generated total, neuronal, and astrocytic MAGL knockout mice by crossing with sox2-, syn1-,and gfap-cre mice.While repeated mild closed head injury results in chronic neuroinflammation,increased expression of TAR DNA-binding protein 43 and phosphorylated tau, neurodegeneration,deteriorations in the synaptic integrity and longterm synaptic plasticity, as well as cognitive decline in wild-type mice, TBI-induced these changes are diminished in total knockout and astrocytic knockout mice.However, these neuroprotective effects were not seen in neuronal knockout mice(Hu et al., 2022), suggesting that previously observed neuroprotective effects produced by pharmacological inactivation of MAGL results primarily from limiting 2-AG degradation in astrocytes, rather than in neurons (Chen et al.,2012; Piro et al., 2012; Zhang et al., 2014, 2015;Mayeux et al., 2017; Hashem et al., 2021; Hu et al.,2022).Moreover, this study provides evidence that expression of MAGL in astrocytes is significantly increased in the hippocampus following TBI,suggesting that 2-AG degradation in astrocytes is likely escalated, which promotes TBI-induced neuroinflammation and neuropathology (Hu et al.,2022).Given the fact that 2-AG possesses profound anti-inflammatory properties (Du et al., 2011; Zhang et al., 2014), the authors conducted a single cell transcriptomic analysis in astrocytes and microglial cells.Interestingly, TBI upregulates the genes associated with immune and inflammatory responses and downregulates the genes linked to anti-inflammatory and maintenance of homeostasis in astrocytes and microglial cells from wild-type and neuronal knockout mice.However,these changes do not occur in astrocytic knockout mice, indicating that limiting 2-AG breakdown in astrocytes enhances resilience to TBI-induced neuroinflammation by suppression of the genes associated with inflammation and enhancement of the genes associated with anti-inflammation or maintenance of the brain homeostasis.In addition, changes in expression of the genes in both astrocytes and microglia and reactivity of astrocytes and microglia suggest that resolving inflammatory responses by 2-AG signaling is controlled by MAGL in astrocytes and that astrocytic 2-AG as a signaling molecule interacts with microglia to decline microglia-mediated inflammatory responses (Figure 1).
What are the downstream signaling molecules in mediating astrocytic MAGL inactivation-produced neuroprotection following TBI? Previous studies showed that 2-AG in protecting the brain from TBI is mediated via CB1 receptors (Panikashvili et al.,2001, 2005).To assess whether MAGL inactivationproduced neuroprotection in TBI is mediated via CB1 receptors, the authors used CB1 receptor knockout mice treated with JZL184, a potent and irreversible inhibitor for MAGL.While JZL184 still exerts protective effects in wild-type mice with TBI, it fails to prevent TBI-induced neuropathology and cognitive decline in CB1 receptor knockout mice (Hu et al., 2022), indicating that CB1 receptors are a downstream molecule of 2-AG in producing neuroprotection against brain trauma.It has been shown that inflammatory prostaglandins are largely derived from 2-AG synthesized in astrocytes, but not in neurons (Viader et al., 2015).Reduced eicosanoids by inactivation of astrocytic MAGL in astrocytic knockout mice might be one of the mechanisms underlying anti-inflammatory and neuroprotective effects in TBI.However, the MAGL inactivation-produced effects are lost in CB1 receptor knockout mice, suggesting that the major neuroprotective effects of inactivation of MAGL in TBI result from 2-AG-mediated signaling.What is the downstream signaling of CB1 receptors?Earlier studies have demonstrated that peroxisome proliferator-activated receptor γ (PPARγ) is a target of 2-AG in resolving neuroinflammation and neuroprotection (Du et al., 2011; Zhang et al., 2014), suggesting that 2-AG is an endogenous PPARγ agonist (Du et al., 2011; Zhang et al., 2014;Hu et al., 2022).PPARγ is an important nuclear receptor functioning as a transcriptional factor, and it displays anti-inflammatory, and neuroprotective properties, which are largely through interacting with nuclear factor kappa B (NF-κB) to suppress NF-κB-mediated transcription and expression of genes involved in neuroinflammation and neurodegeneration.In wild-type and neuronal knockout mice, TBI induces a CB1 receptordependent downregulation of PPARγ expression,while it upregulates phosphorylated NF-κB.However, this does not occur in total knockout and astrocytic knockout mice.These data provide evidence that enhancement of 2-AG signaling by inactivation of MAGL in astrocytes increases expression and activity of PPARγ,which inhibits NF-κB activity (Figure 1).This is true as knocking PPARγ down in astrocytes using the adeno-associated viruses-mediated gene silencing technique diminishes or attenuates the neuroprotection and cognitive improvement in astrocytic knockout mice, suggesting that PPARγ is a key downstream signaling molecule that mediates astrocytic MAGL inactivation produced beneficial effects against brain trauma.This assumption is further supported by the results from the experiments where human PPARγ is overexpressed in astrocytes.Overexpression of PPARγ in astrocytes significantly reduces TBI-induced neuropathology and deterioration in learning and memory in wild-type mice.The results from this study reveal molecular signaling mechanisms by which augmentation of 2-AGsignaling by inactivation of MAGL in astrocytes mitigates TBI-induced neuroinflammation and neuropathology and prevents deterioration in synaptic and cognitive functions (Figure 1).
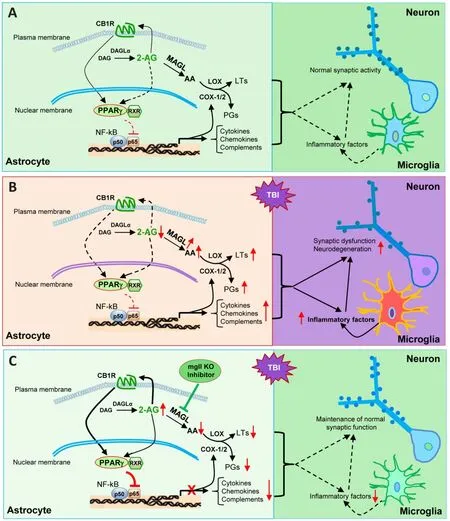
Figure 1 | Cartoon illustrating signaling pathways that mediate astrocytic MAGL inactivation-produced neuroprotection in TBI.
Since 2-AG displays profound anti-inflammatory and neuroprotective properties, enhancement of 2-AG signaling by inactivation MAGL might provide a resolution to terminate inflammatory responses, thereby preventing or treating brain disorders.Thus, MAGL has been proposed as a therapeutic target for neurodegenerative diseases(Chen et al., 2012; Zhang et al., 2014, 2015;Hashem et al., 2021).However, based on the findings by Hu and colleagues, restraining 2-AG degradation by inactivation of MAGL in astrocytes,but not in neurons, produces neuroprotective effects in TBI.In particular, selective inactivation of MAGL in neurons impairs spatial learning and memory (Hu et al., 2022), indicating that proper 2-AG metabolism in neurons is important for maintaining the normal cognitive function.From this point of view, global inactivation of MAGL might not be an optimal approach to achieve ideal therapeutic goals for neurodegenerative diseases as this will disrupt 2-AG degradation in neurons,hence deciphering the cell type-specific role of 2-AG metabolism will enable us to develop a better therapeutic strategy for neurodegenerative diseases, including TBI.
This work was supported by National Institutes of Health grants No.R01NS076815, R01MH113535,and R01AG058621.
Chu Chen
Department of Cellular and Integrative Physiology,Long School of Medicine, University of Texas
Health Science Center at San Antonio, San Antonio,TX, USA
*Correspondence to:
Chu Chen, PhD,chenc7@uthscsa.edu or chen502@gmail.com.https://orcid.org/0000-0003-1287-8059(Chu Chen)
Date of submission:
June 8, 2022Date of decision:
July 12, 2022Date of acceptance:
July 30, 2022Date of web publication:
October 10, 2022https://doi.org/10.4103/1673-5374.355755
How to cite this article:
Chen C (2023)
Endocannabinoid control of neuroinflammation in traumatic brain injury by monoacylglycerol lipase in astrocytes.Neural Regen Res 18(5):1023-1024.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build
upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
- 中国神经再生研究(英文版)的其它文章
- Patient-specific monocyte-derived microglia as a screening tool for neurodegenerative diseases
- Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases
- Inflammation in diabetic retinopathy: possible roles in pathogenesis and potential implications for therapy
- Targeting the nitric oxide/cGMP signaling pathway to treat chronic pain
- Neurosteroids as stress modulators and neurotherapeutics: lessons from the retina
- Myelinosome organelles in pathological retinas:ubiquitous presence and dual role in ocular proteostasis maintenance