
Neuroinflammation and glial activation in the central nervous system: a metabolic perspective
2022-09-27 09:44AssuntaVirtuosoCiroDeLucaSohaibAliKoraiMichelePapaGiovanniCirillo
中国神经再生研究(英文版) 2023年5期
Assunta Virtuoso, Ciro De Luca, Sohaib Ali Korai, Michele Papa, Giovanni Cirillo
Decades of research in glial biology have investigated mechanisms of neuro-glial interplay,demonstrating that neurons and glia intimately cooperate for energy metabolism in the central nervous system (CNS) (Magistretti and Allaman,2018).As neurons invest their adenosine triphosphate in the neurotransmission, glial cells work to support neurons, providing metabolic substrates.Astrocytes, in particular, contribute largely to energy metabolism in CNS: they sense and shuttle nutrients (e.g., glucose and lipids) and hormones (e.g., insulin), accumulate and utilize glycogen for brain demands, influencing their own metabolism and neuronal activity by lactate transport (astrocyte-neuron lactate shuttle).
Alterations of metabolic pathways have been found in glial cells in aging and disease, in particular in astrocytes, and correlated with aberrant inflammatory signaling (Xiong et al.,2022).The consequent activation of glial cells and glial dysfunction (gliopathy) produce first neuroinflammation, maladaptive plasticity,neuronal failure, and later neurodegeneration(Cragnolini et al., 2020).However, systemic lipid and glucose metabolism have not been fully investigated and are essential to understanding both CNS physiological and pathological processes.
Lipid metabolism and neuro-glia homeostasis:
Oxidation of fatty acids contributes to 20% of brain energy demands and occurs primarily in astrocytes (Ioannou et al., 2019).Fatty acids are constituents of the cell membrane phospholipids and are significantly involved in the neuron-glial interaction and neural homeostasis.For example,astrocytes provide fatty acids within lipid droplets during energy deficits and protect hyperactive neurons by detoxifying their toxic fatty acids.In vivo
andin vitro
studies have shown that increased neuronal activity induces the production and accumulation of fatty acids and peroxidized lipids in neurons.To prevent the toxic accumulation of fatty acids, neurons transfer fatty acids to astrocytes via apolipoprotein E lipid particles and convert fatty acids into lipid droplets which are stored in the cells, processed by lipolysis and oxidized in mitochondria.This process, however,leads to increased the production of mitochondrial reactive oxygen species that astrocytes compensate for by upregulating oxidative stressand lipid metabolism-related genes, involved in the protection from free fatty acids toxicity (Gpx8),neutralizing superoxide radicals (Sod1 and Sod3),hydrogen peroxide (Cat), fatty acids transport(Fabp5 and Fapb7) and metabolism (Acsbg1 and Dbi).Accumulating fatty acids in astrocytes, in contrast, progressively lead to oxidative stress(due to mitochondrial failure), cellular/synaptic dysfunction, neuroinflammation, and, lately, cell death.Lipid-driven neuroinflammatory response:
In pathological conditions, astrocytes respond rapidly to perturbation of brain homeostasis(stress, injury, stroke) by changing their morphofunctional features and becoming reactive astrocytes.Apart from the peculiar pattern of gene activation and protein expression [i.e., glial fibrillary acidic protein (GFAP)], reactive astrocytes show increased oxidation of fatty acids as a compensatory mechanism to protect neurons against fatty acids toxicity and provide energy support to neurons.This beneficial phenotype of reactive astrocytes is in contrast with a neurotoxic phenotype, expressing high levels of fatty acids binding proteins, and characterized by increased oxidation of fatty acids, reactive oxygen species production, and release of saturated lipids that induce cell death (Guttenplan et al., 2021).Alteration of lipid metabolism is associated with dysregulation of the inflammaraft, an enlarged lipid raft with adaptor molecules and activated receptors, such as toll-like receptor 4 (TLR4),that promotes inflammatory signaling (Miller et al., 2020).Activation of TLR4 signaling is related to receptor dimerization that, in turn,is dependent upon the cholesterol membrane content of the lipid rafts (Figure 1).Evidence has demonstrated that depletion of cholesterol prevents lipopolysaccharide-induced TLR4 dimerization whilst increased cholesterol in the lipid rafts facilitates TLR4 dimerization.Following TLR4 activation, highly expressed in microglia, downstream nuclear factor kappa B activates caspase 1 resulting in the release of cytokines, chemokines, and lipids triggering a neuroinflammatory reaction.Apart from TLR4,other receptors and inflammaraft-dependent pathways activate inflammatory mechanisms.For example, after nerve injury, interferons γ activate Janus kinase and activators of transcription 3 that induce microglial activation via purinergic P2X4 receptor.Interestingly, cholesterol regulates purinergic P2X4 receptor activity and its membrane localization is regulated by the association with lipid rafts or through vesicle exocytosis (Miller et al., 2020).
Modulation of TLR4 dimerization by apolipoprotein A-I binding protein has been proven effective in reducing neuroinflammation and glial activation(Woller et al., 2018).Apolipoprotein A-I binding protein binds to apolipoprotein A-I and highdensity lipoprotein and causes cholesterol efflux from cells via adenosine triphosphate-binding cassette protein A1 and G1to high-density lipoprotein (cholesterol, sphingomyelin, and phosphatidylcholine via adenosine triphosphatebinding cassette protein G1) and apolipoprotein A-I (cholesterol and phosphatidylcholine via adenosine triphosphate-binding cassette protein A1).Cholesterol efflux compromises the integrity of lipid raft, inhibits the inflammatory TLR4 signaling preventing its dimerization, and increases the expression and activity of adenosine triphosphate-binding cassette protein A1 and G1,potentially mitigating the inflammatory response.
Among the other targets, delicate, a protein associated with familial Parkinson’s disease, plays a crucial role in supporting lipid rafts integrity but also in the negative feedback mechanisms that suppress glial activation (Miller et al., 2020).Lack of deglycase changes flotillin-1 stability,altering cellular cholesterol levels, membrane fluidity, and lipid raft-dependent endocytosis but also impairs glutamate uptake due to reduced expression of glutamate transporter 2 (Figure 1).These mechanisms further deteriorate fatty acid regulation in astrocytes and impair the inflammaraft stability.Additionally, deglycase is involved in the termination of TLR4 signaling via receptor endocytosis and the inhibition of the interferons γ inflammatory response.
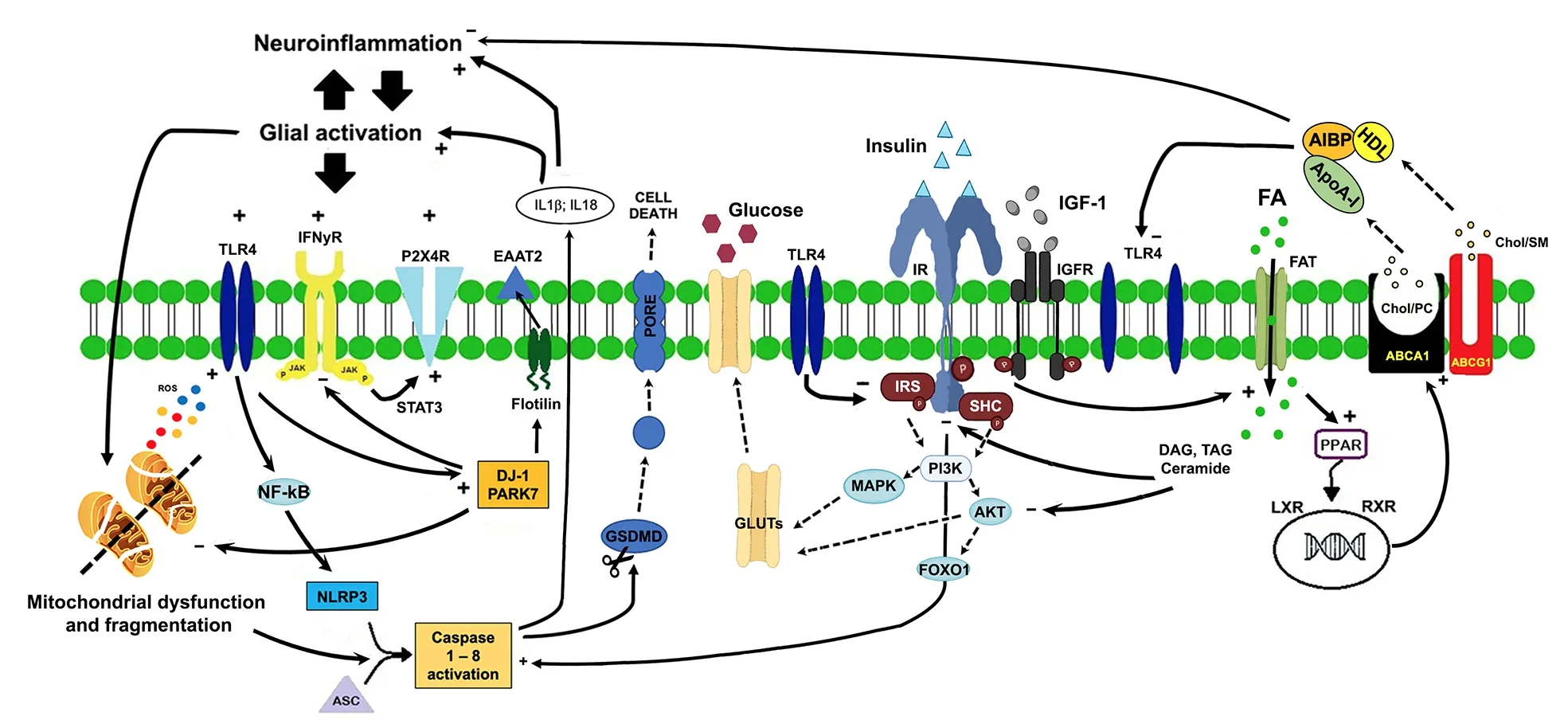
Figure 1 | Lipid-glucose metabolic changes following neuroinflammation and glial activation.
A link between lipid metabolism and neuroinflammation is represented by inflammasomes, macromolecular complexes that trigger a neuroinflammatory response.NOD-like receptor P3 (NLRP3) is the most widely studied in CNS and responds to the greatest variety of endogenous and exogenous stimuli, including reactive oxygen species and fatty acids.Activation of the NLRP3 inflammasome complex (ASC,NLRP3, and caspase-1) allows the cleavage of pro-caspase-1 into its active isomer caspase-1, which produces active pro-inflammatory interleukin-1β and interleukin-18, that ultimately leads to pyroptosis.
Extracellular apolipoprotein E levels are under the control of microglial mediators and cholesterol metabolism in astrocytes (Qi et al., 2021).Therefore, apolipoprotein E represents a molecular bridge between neuroinflammation and lipid metabolism in healthy conditions and diseases.In particular, the apolipoprotein E ɛ4 isoform, known to be the strongest genetic risk factor for the lateonset form of Alzheimer’s disease, disrupts brain mitochondrial function and neuron-astrocyte coupling of fatty acids metabolism, impairing the bioenergetic and synaptic support of astrocyte to neurons and suggesting a neuronal mitochondrial dysfunction via inter-cellular coupling of lipid metabolism.
Glucose metabolism in neuro-glial homeostasis:
The major source of energy in CNS depends upon glucose metabolism through three principal metabolic pathways: glycolysis, tricarboxylic acid cycle, and mitochondrial oxidative respiration(Bonvento and Bolaños, 2021).Glucose uptake in the brain passes first through the endothelium via glucose transporter 1 (GLUT1) (55 kDa), then is transported into astrocytes by GLUT1 (45 kDa),and finally is transferred to neurons via neuronal GLUTs (Yin et al., 2016).The preferential glucose metabolism in astrocytes is the glycolytic activity,as also demonstrated by the low expression of tricarboxylic acid and mitochondrial enzymes.Astrocytes and neurons are metabolically coupled to face the energy needs of neurotransmission,preserve the redox balance, and modulate the neurotransmitter-receptor activity.Intermediate metabolites of the astrocytic glycolysis (lactate and serin) are shuttled to neurons for mitochondrial oxidation and biosynthesis of other products.Glucose disposal is related to the activity of insulin and insulin-like growth factors 1 and 2 that show neurotrophic and neuroprotective effects through insulin receptors activity.Insulin/insulin-like growth factor receptors are widely expressed in the CNS onto neurons, astrocytes, and microglial cells and are active after phosphorylation of tyrosine kinase/insulin receptor substrate proteins.Phosphorylation of insulin receptor substrate proteins activates the phosphatidylinositol 3-kinase and the phosphoinositide-dependent protein kinase 1 that phosphorylate the protein kinase B that regulates several target proteins of the insulin/IGF signaling pathway, such as the transcription factor forkhead box O1, that regulates several neuronal functions such as neurite outgrowth, neuronal survival, N-methyl-D-aspartate, alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid, and gamma-aminobutyric acid receptor expression and localization and,finally, glucose uptake (Scherer et al., 2021).In particular, activation of the PI3K-MAPK-AKT pathway leads to GLUT4 translocation to the cell membrane, thereby increasing glucose uptake(Figure 1).In this way, glucose can be converted to pyruvate by glycolysis and subsequently pass into the mitochondria and change into acetyl-CoA for the Krebs’ cycle.
Activation of astrocytes requires metabolic reprogramming and switching to meet the increased energy demands and/or functional alterations in response to injury or stress.However, upregulation of glycolysis produces more lactate to support neuronal energy metabolism in response to the enhanced brain activity but also accelerates inflammation by fueling the astrocyte activation.In this light, therefore, astrocytic glycolysis represents a double-edged sword for the development of neuro-inflammatory disorders.
Altered glucose metabolism in CNS disorders:
Altered glucose transports and metabolism,together with dysregulation of neuronal/astrocytic GLUTs and brain insulin resistance,occur in physiological aging, neuroinflammatory and neurodegenerative disorders (Scherer et al.,2021).Compromised glucose metabolism is the signature of normal brain aging: brain glucose uptake inversely correlates with the decline of insulin-sensitive and expression of vascular (GLUT1 55KDa) and neuronal (GLUT3, GLUT4) GLUTs.Interestingly, the expression of the astrocytic GLUT1 (45 kDa) increased with age, suggesting a metabolic shift in neurons and astrocytes (Yin et al., 2016).In experimental models of multiple sclerosis, an increased glycolytic metabolism supports the transmigration of brain-infiltrating macrophages, boosting neuroinflammatory reaction (Kaushik et al., 2019).Glucose metabolism in Alzheimer’s disease has been associated with insulin resistance,deficiency of insulin and its receptors, and/or dislocation of the GLUTs, leading to abnormal tau phosphorylation and neurofibrillary degeneration(Yin et al., 2016).Insulin resistance also leads to decreased phosphorylation of insulin receptor substrate and AKT signaling that reduces the inhibitory response to forkhead box O1 protein,resulting in an increased forkhead box O1 expression.Moreover, it has been demonstrated that insulin resistance in diabetes is tightly correlated with an increased risk of Alzheimer’s disease because cortical and hippocampal neurons are more vulnerable to Aβ and tau toxicity.Accordingly, evidence has suggested that insulin resistance accelerates neuronal dysfunction depriving neurons of the neurotrophic properties of insulin and making them more susceptible to external injurious stimuli.These modifications lead to alteration of the neuroglial synaptic homeostasis, an increase of lipid peroxidation, and oxidative stress that ultimately lead to mitochondrial dysfunction (increased reactive oxygen species production, alteration of mitochondrial membrane depolarization,decreased Oconsumption, COproduction, and loss of adenosine triphosphate content), energy deficit and cell death.
Conclusion:
Dysregulation of lipid and glucose metabolism is associated with impaired neuron-toglia crosstalk, neuroinflammation, and activation of glial cells (reactive gliosis).Metabolic changes also reflect morphological alteration in reactive astrocytes, whose morphological feature is the increased expression of GFAP, the epoxidation and hyperpalmitoylation of GFAP cysteine residues contribute to GFAP disruption and aggregation(DeLuca et al., 2022), boosting an inflammatory response.Increased energy demand and an altered, inappropriate metabolic response could be detrimental to neuron-glia homeostasis,paving the way for neurodegeneration.Therefore,understanding and targeting metabolic changes in a neuron-glia interplay might represent an increasing research field at the base of promising treatments for neuroinflammatory and neurodegenerative disorders.This work was supported by grants from Regione Campania (L.R.N.5 Bando 2003 to MP), the Italian Minister of Research and University (PRIN 2007 to MP; PRIN 2017-2017XJ38A4_003 to GC and MP)and UNIMIB (Progetto ID 2019-ATESP-0001 and Progetto ID 2018-CONV-0056 to AV).
Assunta Virtuoso, Ciro De Luca,Sohaib Ali Korai, Michele Papa,Giovanni Cirillo
Division of Human Anatomy, Laboratory of Morphology of Neuronal Networks and Systems Biology, Department of Mental and Physical Health and Preventive Medicine, University of Campania“Luigi Vanvitelli”, Naples, Italy
#These authors contributed equally to this work.
*Correspondence to:
Giovanni Cirillo, MD, PhD,giovanni.cirillo@unicampania.it.https://orcid.org/0000-0003-3183-8773(Giovanni Cirillo)
Date of submission:
May 23, 2022Date of decision:
July 20, 2022Date of acceptance:
August 3, 2022Date of web publication:
October 10, 2022https://doi.org/10.4103/1673-5374.355754
How to cite this article:
Virtuoso A, De Luca C,Korai SA, Papa M, Cirillo G (2023)Neuroinflammation and glial activation in the central nervous system: a metabolic perspective.Neural Regen Res 18(5):1025-1026.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
- 中国神经再生研究(英文版)的其它文章
- Patient-specific monocyte-derived microglia as a screening tool for neurodegenerative diseases
- Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases
- Inflammation in diabetic retinopathy: possible roles in pathogenesis and potential implications for therapy
- Targeting the nitric oxide/cGMP signaling pathway to treat chronic pain
- Neurosteroids as stress modulators and neurotherapeutics: lessons from the retina
- Myelinosome organelles in pathological retinas:ubiquitous presence and dual role in ocular proteostasis maintenance