
Neutrophil-derived interleukin-17A participates in neuroinflammation induced by traumatic brain injury
2022-09-27 09:44XiaoJianXuQianQianGeMengShiYangYuanZhuangBinZhangJinQianDongFeiNiuHaoLiBaiYunLiu
中国神经再生研究(英文版) 2023年5期
Xiao-Jian Xu, Qian-Qian Ge, , Meng-Shi Yang, , Yuan Zhuang, , Bin Zhang, , Jin-Qian Dong, , Fei Niu, Hao Li, , Bai-Yun Liu, ,
Abstract After brain injury, infiltration and abnormal activation of neutrophils damages brain tissue and worsens inflammation, but the mediators that connect activated neutrophils with neuroinflammation have not yet been fully clarified.To identify regulators of neutrophil-mediated neuroinflammation after traumatic brain injury, a mouse model of traumatic brain injury was established by controlled cortical impact.At 7 days post-injury (sub-acute phase), genome-wide transcriptomic data showed that interleukin 17A-associated signaling pathways were markedly upregulated, suggesting that interleukin 17A may be involved in neuroinflammation.Double immunofluorescence staining showed that interleukin 17A was largely secreted by neutrophils rather than by glial cells and neurons.Furthermore, nuclear factor-kappaB and Stat3, both of which are important effectors in interleukin 17A-mediated proinflammatory responses, were significantly activated.Collectively, our findings suggest that neutrophil-derived interleukin 17A participates in neutrophil-mediated neuroinflammation during the subacute phase of traumatic brain injury.Therefore, interleukin 17A may be a promising therapeutic target for traumatic brain injury.
Key Words: immune infiltration; innate immunity; interleukin-17A; neurodegenerative disease; neuroinflammation; neutrophils; secondary brain injury;transcription factor; transcriptome; traumatic brain injury
Introduction
Traumatic brain injury (TBI) leads to devastating cerebral functional impairment and cognitive deficits (Rabinowitz and Levin, 2014) and is considered to be a crucial risk factor for neurodegenerative disease(Arena et al., 2020; Xu et al., 2021).Cumulative evidence suggests that neuroinflammation, elicited by multiple signaling cascades such as interferon-β and the complement system, is strongly associated with longterm cognitive decline following TBI (Barrett et al., 2020; Alawieh et al.,2021; Iacono et al., 2021).However, the fundamental drivers underlying TBI-induced neuroinflammation remain largely unknown, which has hampered the development of target-specific treatment approaches.
Neutrophils, the most abundant circulating leukocytes, are crucial effector cells of the innate immune system (Papayannopoulos, 2018).Neutrophils exert critical effector functions that contribute to the host defense primarily through phagocytosis, degranulation, and the formation of neutrophil extracellular traps (NETs) (Gierlikowska et al., 2021).However, accumulating evidence indicates that infiltration and abnormal activation of neutrophils not only cause tissue damage and amplify inflammation (Wang, 2018; Lehman and Segal, 2020), but also contribute to chronic sterile inflammation and activation of the adaptive immune response (Németh et al., 2020; Rosales,2020).Moreover, there is emerging evidence of roles for neutrophils in neuroinflammation and neurodegenerative disease, including Alzheimer’s disease (Zenaro et al., 2015) and TBI (Vaibhav et al., 2020).Although infiltrated neutrophils have been implicated in TBI-induced secondary injury,the mediators that bridge activated neutrophils and neuroinflammation have not yet been fully elucidated.Therefore, the purpose of the current study was to identify regulatory elements that account for neutrophil-mediated neuroinflammation after TBI.
Methods
Animals
Adult female and male C57BL/6 mice (specific-pathogen-free, 25–30 g, 8–10 weeks old) were purchased from Beijing Vital River Experimental Animals Technology Co., Ltd.(Beijing, China; license No.SCXK (Jing) 2016-0006) and allowed to acclimate to the housing facility for at least 7 days prior to any experimental procedures.Mice of both sexes were included in approximately equal numbers (~1:1) in the present work.Mice were housed in a controlledtemperature (22 ± 2°C) and humidity-barrier facility (50–60%) with food and water available ad libitum on a 12/12-hour light/dark cycle.Up to five mice were housed in each cage.Animal procedures were reviewed by the Beijing Neurosurgical Institute Animal Care and Use Committee (approval No.202101018) and approved on April 22, 2021.All experiments were designed and are reported according to the Animal Research: Reporting ofIn Vivo
Experiments (ARRIVE) guidelines (Kilkenny et al., 2010).A flow chart of the experimental procedures is shown in Figure 1.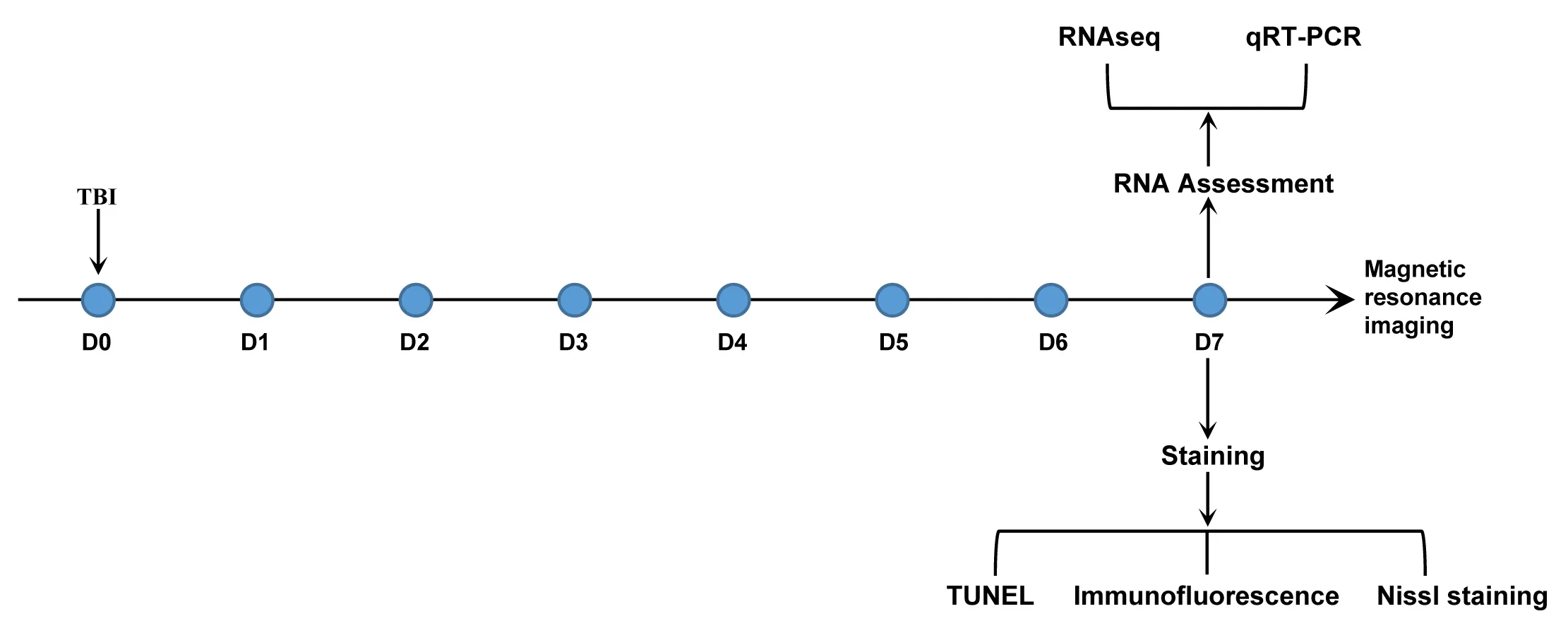
Figure 1|Flow chart of the experimental design.
C ontrolled cortical impact model
The mice were randomly assigned to the control group or the TBI group (n
= 35 per group).A controlled cortical impact (CCI) model was established,as we described previously (Zhang et al., 2020).Briefly, mice, anesthetized with 2% isoflurane (RWD Life Science Co., Shenzhen, China) by inhalation were placed in a stereotaxic frame (RWD Life Science Co.).Body temperaturewas maintained at 37.0 ± 0.5°C with a heating pad throughout the surgical procedure.A craniotomy (~4.0-mm diameter) was performed on the right parietal cortex (Xu et al., 2022) without damaging the dura mater.Then, CCI injury was induced with a 3-mm impactor tip using an electromagnetic CCI device (Pinpoint PCI3000 Precision Cortical Impactor, Hatteras Instruments,Cary, NC, USA).The following parameters were used: velocity 3 m/s; dwelltime 20 ms; and impact depth 1.5 mm.After the impact, bone wax was used to seal the burr hole, followed by suturing of the scalp incision.Magnetic resonance imaging
Because 7 days post-injury (dpi) is considered to be the subacute phase of TBI (Rubovitch et al., 2011; Meng et al., 2017; Vinh To et al., 2022), 7 dpi was chosen as the target time point.At 7 dpi, mice were anesthetized with 3%isoflurane and maintained under anesthesia with 1–2% isoflurane.Then, MRI was performed using a 7.0 Tesla small-bore animal scanner (Bruker Biospec,Ettlingen, Germany).The mouse head was immobilized with custom-made head holder device, and respiration was monitored with a small-animal monitoring system (SA Instruments, Inc., New York, NY, USA) throughout the scan procedure.T2-weighted images were acquired with following parameters: repetition time = 2795 ms; echo time = 35 ms; echo spacing =11.667 ms; thickness = 0.5 mm; field of view = 22 mm × 22 mm.
Brain tissue collection
At 7 dpi, after being deeply anesthetized with isoflurane, mice were transcardially perfused with ice-cold 0.9% saline solution.For RNA sequencing and real-time polymerase chain reaction (qPCR), the brains were removed, and cerebral perilesional cortex tissues were collected on a chilled stainless steel plate placed on a bed of crushed ice.RNA was obtained from the cortices using TRIzol reagent (Invitrogen, Carlsbad, CA,USA) following the manufacturer’s instructions.For immunofluorescence,terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL),and Nissl staining, saline perfusion was followed by perfusion with 4%paraformaldehyde in phosphate-buffered saline (PBS).Then, the brains were collected and post-fixed in 4% paraformaldehyde overnight at 4°C.After post-fixation, the brains were cryoprotected in 30% sucrose for 48 hours at 4°C.Finally, the brains were embedded in optimum cutting temperature compound (Sakura Finetek USA, Inc., Torrance, CA, USA), and 20-μm coronal sections were made using a Leica cryostat (Leica Biosystems Nussloch GmbH,Nussloch, Germany).
Immunofluorescence and Nissl staining
To assess structural integrity, cellular identity, and cell state,immunofluorescence and Nissl staining were used.Following washing with PBS, a 37°C slide warmer (Leica Biosystems Nussloch GmbH) was used to dry the frozen sections.Subsequently, the sections were subjected to heatinduced antigen retrieval in 10 mM sodium citrate buffer.The sections were then permeabilized and blocked with 5% bovine serum albumin (Merck KGaA, Darmstadt, Germany), 0.3% Triton X-100 in PBS for 1 hour at ambient temperature (23–25°C).Afterwards, the sections were incubated with primary antibodies overnight at 4°C.For single immunofluorescence, after washing,corresponding secondary antibodies were added to the slides, which were then incubated for 1 hour at ambient temperature (23–25°C).For double immunofluorescence, the slides were incubated overnight with primary antibodies at 4°C followed by incubation with secondary antibodies for 1 hour at ambient temperature (23–25°C).Finally, the nuclei were counterstained with 4′,6-diamidino-2-phenylindole, and coverslips were mounted on the slides using Fluoromount-G medium (Thermo Fisher Scientific Inc., Waltham,MA, USA).The primary antibodies used were as follows: rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1; 1:500, Wako, Osaka, Japan, Cat#019-19741, RRID: AB_839504), goat anti-Iba1 (1:200, Novus Biologicals,Centennial, CO, USA, Cat# NB100-1028, RRID: AB_521594), mouse anti-glial fibrillary acidic protein (GFAP; 1:100, Cell Signaling Technology, Danvers, MA,USA, Cat# 3670, RRID: AB_561049), rabbit anti-IL-17A (1:100, Proteintech,Wuhan, China, Cat# 26163-1-AP, RRID: AB_2880409), mouse anti-neuronal nuclear protein (NeuN; 1:500, Abcam, Cambridge, UK, Cat# ab104224, RRID:AB_10711040), rabbit anti-signal transducer and activator of transcription factor (Stat)1 (1:100 Proteintech, Cat# 10144-2-AP, RRID: AB_2286875), rabbit anti-Stat3 (1:100, Proteintech, Cat# 10253-2-AP, RRID: AB_2302876), goat anti-S100A8 (1:100, Novus Biologicals, Cat# AF3059, RRID: AB_2184254), and rabbit anti-nuclear factor kappa-B (NF-κB)phophoS536 (1:400, Cat# ab86299,Abcam, RRID: AB_1925243).Donkey anti-goat-Cy3 (Cat# 705-165-003, RRID:AB_2340411)/mouse-Cy2 (Cat# 715-225-150, RRID: AB_2340826)/rabbit-Cy3(Cat# 711-165-152, RRID: AB_2307443) secondary antibodies were purchased from Jackson ImmunoResearch Inc.(PA, USA).The slides were observed using s Nikon Instruments A1 confocal laser microscope (Nikon, Tokyo, Japan).
For Nissl staining, brain sections were first rinsed with distilled water for 2 minutes, and then stained with cresyl violet (Cat# C0117, Beyotime, Shanghai,China) for 5 minutes at 50°C.After washing with distilled water, sections were dehydrated with 95% ethanol and cleared in xylene, followed by mounting with DPX mounting medium (Merck KGaA).The sections were observed using an Aperio AT2 digital pathology scanner (Leica Biosystems Nussloch GmbH).
TUNEL staining
To assess cell death after TBI, TUNEL staining was conducted according the manufacturer’s instructions (In Situ Cell Death Detection Kit, Roche, Basel,Switzerland).Briefly, frozen slides were fixed with 4% paraformaldehyde in PBS at ambient temperature (23–25°C) for 20 minutes.After washing and subsequent permeabilization with a solution containing 0.1% Triton X-100 and 0.1% sodium citrate, the sections were incubated with the TUNEL reaction mixture for 60 minutes at 37°C in the dark.Then, the slides were rinsed with PBS, and coverslips were mounted using Fluoromount-G.The slides were observed using a Nikon Instruments A1 confocal laser microscope.
RNAseq processing and analysis
To investigate the underlying mechanisms that induce secondary brain injury,next-generation sequencing was used.Purified poly (A) RNA was fragmented and then reverse-transcribed to generate a complementary DNA (cDNA)library using an mRNA-Seq sample preparation kit (Illumina, San Diego, CA,USA).The cDNA library, which had an average insert size of 300 ± 50 bp, was paired-end sequenced using an Illumina HiSeq4000 apparatus.After the initial filtering step, HISAT2 (http://www.ccb.jhu.edu/software/hisat/index.shtml)was used to align reads to the mouse genome.Then, gene and transcript expression levels were quantified using StringTie (version 1.3.0; http://ccb.jhu.edu/software/stringtie/).DESeq2 was used to perform differential expression analysis.Genes with |log2(fold change)| > 1 and false discovery rate < 0.05 were designated as differentially expressed genes (DEGs).A mouse transcription factor list, downloaded from The Animal Transcription Factor DataBase (AnimalTFDB; http://bioinfo.life.hust.edu.cn/AnimalTFDB/#!/), was used to identify differentially expressed transcription factors among the DEGs at the mRNA level.
Functional enrichment analysis
Metascape (https://metascape.org/gp/index.html#/main/step1) was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) annotation.Terms with a false discovery rate < 0.05 were defined as significantly enriched terms.
To further understand the pathway alterations vulnerable to perturbation by TBI, gene set variation analysis (GSVA, 1.38.2; https://bioconductor.org/packages/release/bioc/html/GSVA.html) was performed.GSVA is a nonparametric and unsupervised approach that provides more power to detect subtle pathway activity changes than sample-wise enrichment methods(Hänzelmann et al., 2013).Limma package (3.46.0; https://bioconductor.org/packages/release/bioc/html/limma.html) was then used to identify differentially expressed pathways.
Protein-protein interaction networks were generated from the STRING database (https://string-db.org/) after uploading the list of the differentially expressed genes.The Cytohubba application in Cytoscape (https://cytoscape.org/) was then used to identify hub genes via the node degree method.Finally, the interaction network was constructed with Cytoscape.
qPCR
To verify mRNA expression level, qPCR was conducted.After the removal of possible contaminating genomic DNA with DNase I (AM1906, Life Technologies/Ambion, Carlsbad, CA, USA), cDNA was generated with the superscript II reverse transcription system (Invitrogen).LightCycler 480 (Roche)and used to perform qRT-PCR with the following conditions: 40 cycles of 15 seconds at 95°C, 5 seconds at 95°C, and 31 seconds at 60°C.Gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH)expression, and the relative amounts of mRNA were determined by the 2method (Xu et al., 2017).The sequences of the primers used are as follows:Stat1 forward primer 5′-GGA AGG GGC CAT CAC ATT CA-3′; Stat1 reverse primer 5′-TAC TTC CCA AAG GCG TGG TC-3′; Stat3 forward primer 5′-TCC AGT CTG TAG AGC CAT A-3′; Stat3 reverse primer 5′-CGC ATC CAT GAT CTT ATA GC-3′; GAPDH forward primer 5′-GTG GCA AAG TGG AGA TTG-3′; GAPDH reverse primer 5′-GTG GAG TCA TAC TGG AAC A-3′.
Statistical analysis
Sample size determination was based on historical data (Real et al., 2018;Schene et al., 2020; Xu et al., 2022), and no statistical methods were used to predetermine sample size.The investigators were not blinded to the group assignments.Statistical analyses were performed using R statistical software(4.0.5) (https://www.r-project.org/).Data are presented as mean ± standard error of the mean (SEM).Differences were determined by unpaired Student’st
-test and non-parametric Mann-WhitneyU
test for data with normal and non-normal distribution, respectively.A probability value ofP
< 0.05 was taken to be statistically significant.Results
TBI triggers morphological and cytoarchitectural abnormalities and widespread neuroinflammation during the subacute phase
To assess the extent of brain damage caused by CCI at 7 dpi, we first used noninvasive MRI.T2-weighted MRI images showed that the structural integrity of the cortex was destroyed at the impact site, although no prominent midline shift was identified.Moreover, extensive edematous tissues appeared in the ipsilateral hemisphere, expanding to involve the cortex, hippocampus,corpus callosum, and lateral ventricles (Figure 2A).Nissl staining indicated that the most abundant neurons underwent cell death other than cerebral cortical loss at the peri-injury site, and gliosis increased dramatically (Figure 2A).Consistent with this, Iba1 immunolabeling demonstrated that microglia were much more broadly and intensively activated after TBI, not only at the lesion but also in areas distal from the lesion or even in the contralateral hemisphere, indicating widespread neuroinflammation (Figure 2B).In addition, TBI induced significant levels of apoptosis, as evidenced by TUNEL-positivity of different cell types during the subacute phase (Figure 2B).Taken together, these results suggest that TBI evokes morphological and cytoarchitectural abnormalities and widespread neuroinflammation during the subacute phase.
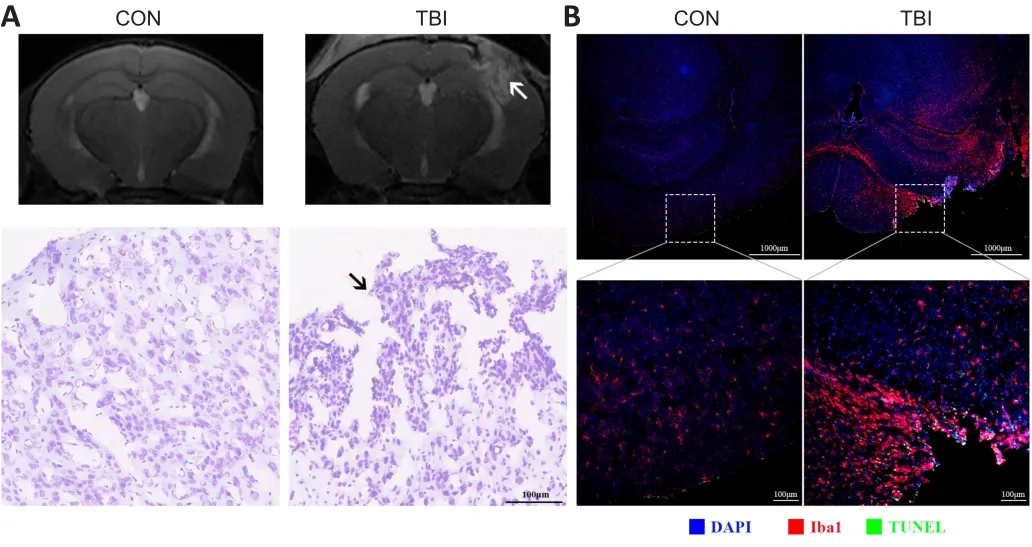
Figure 2 | TBI triggers morphological and cytoarchitectural abnormalities and widespread neuroinflammation at 7 days post-injury.
TBI perturbs the genome-wide transcriptome and upregulates IL-17A-related signaling pathways
Although the mechanisms underlying TBI-induced secondary injury have been studied intensively, the molecular mechanisms governing these processes are not yet completely understood.To explore TBI-associated regulatory cascades, genome-wide transcriptomic profiling of 12 cDNA libraries was conducted.A total of 2138 genes were identified as being differentially expressed at 7 dpi, of which 1973 (92.3%) were upregulated (Additional Figure 1A).Furthermore, DEGs distinguished the TBI group from the control group fairly well, highlighting the substantial biological significance of DEGs in secondary brain injury (Additional Figure 1B).Gene-gene interaction networks are essential for the execution of complex biological processes,and hub genes are thought to have a widespread influence on the overall network.Construction of a protein-protein interaction network revealed that inflammation-associated genes, including tumor necrosis factor alpha (TNFα),Toll-like receptors, STATs, and IL-1β, played a predominant role in the network in the subacute phase (Figure 3A).
To obtain further insight into DEG functions, KEGG analysis was performed.KEGG enrichment analysis demonstrated that innate immune responses were significantly enriched, including the Nod-like receptor signaling pathway,natural killer cell-mediated cytotoxicity, and the complement and coagulation cascades (Figure 3B).As mentioned above, the majority of the DEGs were upregulated, and inflammatory responses dominated the pathological processes, which implied that inflammation, inflammation-induced macrobiomolecular consumption, and energy crisis jointly exacerbated the secondary injury.
KEGG analysis and GSVA both highlighted the massive activation of IL-17A–related signaling pathways in secondary brain injury (Figure 3B–D).IL-17A,which confers protection against pathophysiological insults, recently has attracted more attention in the context of autoimmune and neurological diseases (Ni et al., 2018; McGeachy et al., 2019; McGinley et al., 2020).However, the cellular source of IL-17A and its potential roles in TBI have not yet been not fully elucidated.
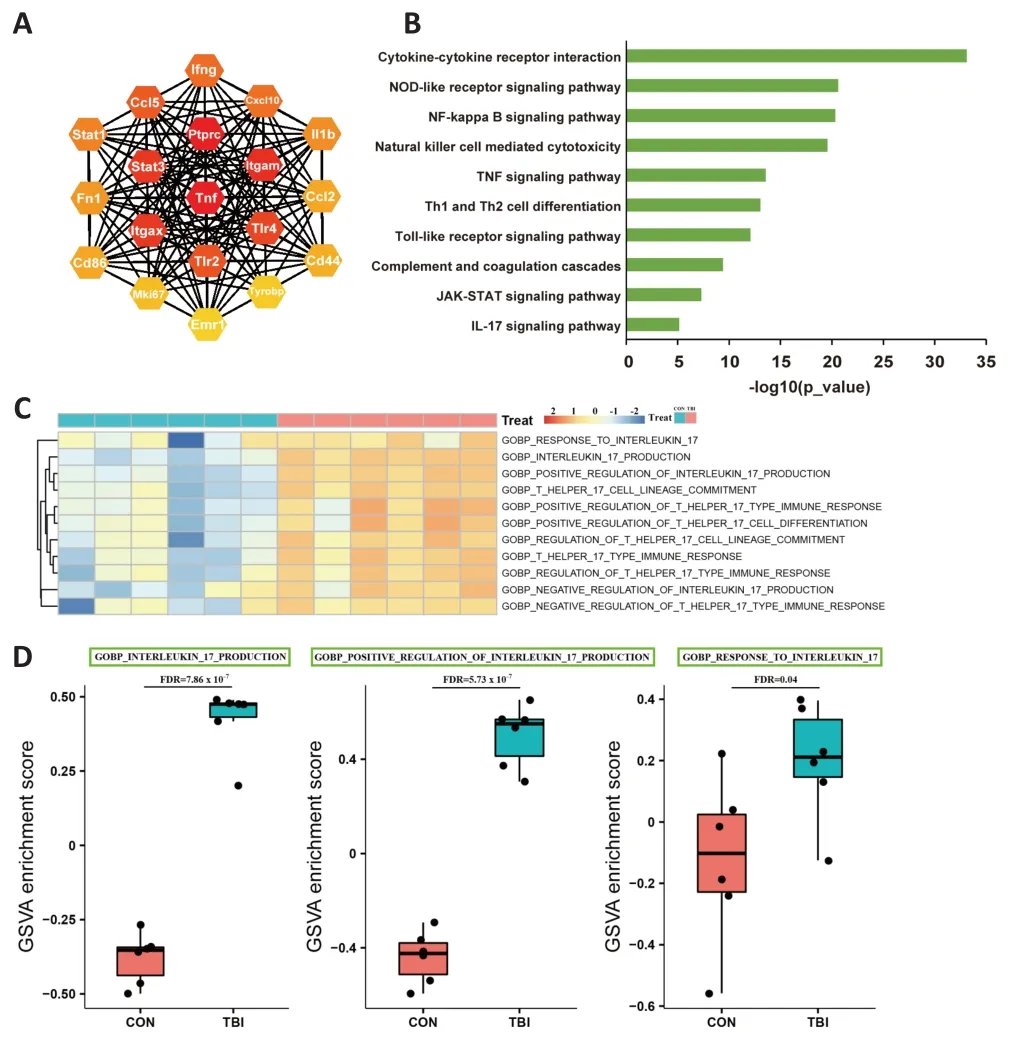
Figure 3 | TBI evokes genome-wide transcriptomic changes and activates IL-17A signaling cascades.
Neutrophils are the main cellular source of IL-17A during the subacute phase
Besides well-known IL-17A–producing cells, such as T helper 17 cells and tissue-resident innate immune cells such as γδ T cells (Monin and Gaffen,2018), emerging evidence shows that glial cells within the central nervous system, including microglial cells and astrocytes, can secrete IL-17A under the physiological and pathophysiological conditions (Kawanokuchi et al., 2008;Zhang et al., 2017; Luo et al., 2019; Di Filippo et al., 2021).To determine the cellular source of IL-17A in the ipsilateral peri-injury cortex during the subacute phase, double immunofluorescence was used to stain cells for IL-17A and one of three other markers: GFAP (astrocyte marker (Alawieh et al.,2021)), Iba-1 (microglia marker (Song et al., 2021)), or NeuN (neuronal marker(Liu et al., 2021)).The results demonstrated that IL-17A did not colocalize with GFAP or NeuN, and only infrequently colocalized with Iba-1 (Figure 4A–C).Given that TBI can facilitate parenchymal neutrophil infiltration (Jassam et al., 2017) and that neutrophils can produce IL-17A in specific disease states (Li et al., 2010; Hu et al., 2017), we speculated that neutrophils might express IL-17A during the subacute period of TBI.S100 proteins are a family of small calcium-binding cytosolic proteins, of which S100A8 and S100A9 are abundantly and constitutively expressed by neutrophils and account for approximately 45% of the cytosolic proteins in neutrophils (Xia et al., 2017;Wang et al., 2018).S100A8/A9 expression was recently used to determine neutrophil identity based on single-cell transcriptome analysis of the mouse brain (Ximerakis et al., 2019).Our immunofluorescence results showed that IL-17A colocalized with S100A8, which, coupled with the shape of the nuclei,implied that neutrophils were the main cellular source of IL-17A during the subacute phase of TBI (Figure 4D).
STATs and NF-κB may be implicated in IL-17A-mediated neuroinflammation
In response to diverse physiological and pathological stimuli, transcription factors fine-tune spatiotemporal gene expression (Shaban and Seeber, 2020).We found that TBI induced differential expression of 118 transcription factors at the mRNA level, 103 of which were upregulated, including STATs (Figure 5A).Both qRT-PCR and immunofluorescence corroborated that mRNA and protein levels of STAT1 and STAT3 were significantly upregulated at 7 dpi(Figure 5A–C).On the one hand, STAT3 is essential for IL-17A production, and on the other hand, IL-17A can promote neuroinflammation through STATs,especially STAT3 (Zong et al., 2014; You et al., 2017).Generally, IL-17A induces a proinflammatory response through the transcription factor NF-κB, whose activity is controlled by phosphorylation (Christian et al., 2016; Chong et al.,2020).Therefore, the levels of phosphorylated NF-κB were determined by immunofluorescence although no change was observed at the mRNA level.The results showed that phosphorylated NF-κB was dramatically elevated at the peri-injury site (Figure 5D).Collectively, these findings suggest that STATs and NF-κB might be involved in IL-17A–mediated neuroinflammation.
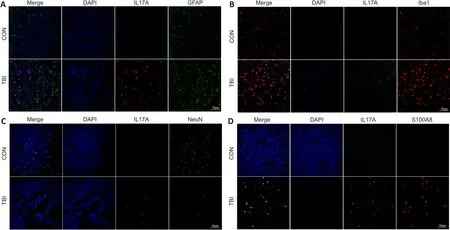
Figure 4|Neutrophils are the main cellular source of IL-17A at 7 dpi.
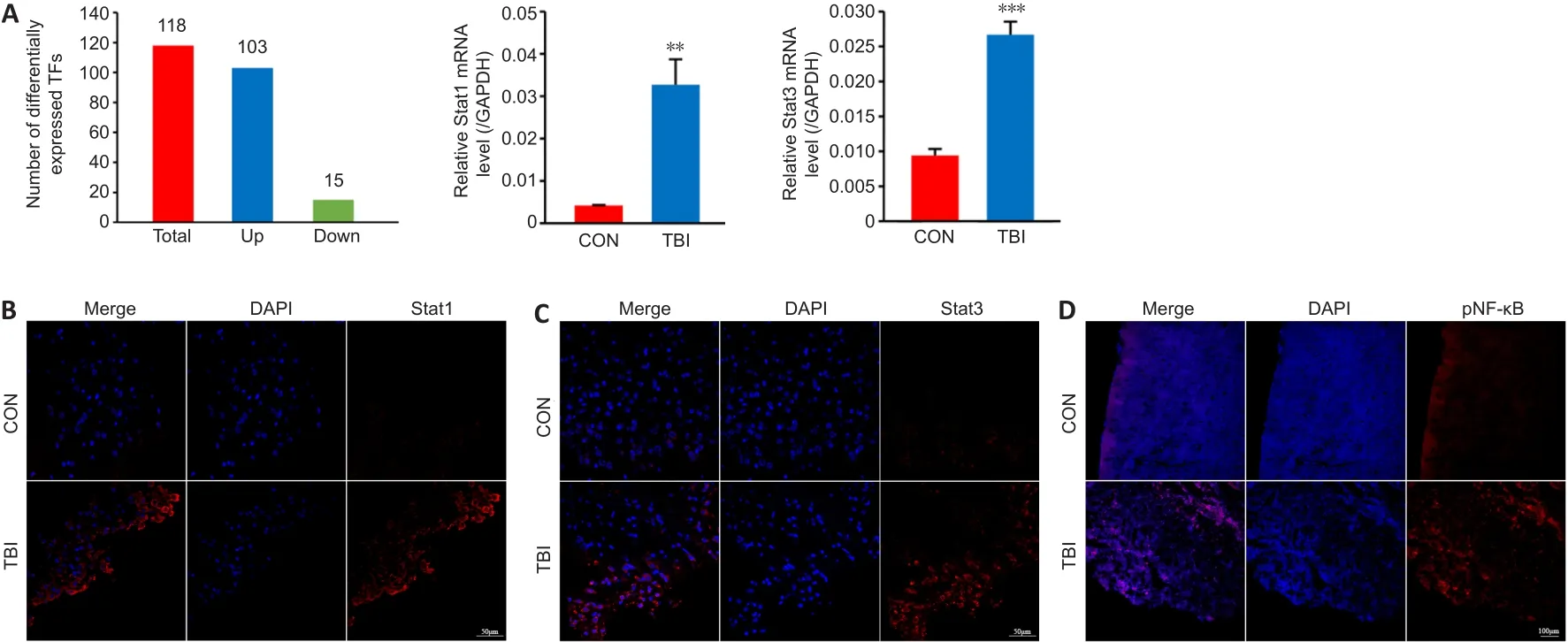
Figure 5 | STATs and NF-κB may be involved in IL-17A-associated neuroinflammation.
Discussion
The innate immune system constitutes the first line of the host’s defense against exogenous and endogenous danger signals (Yang et al., 2021).Neutrophils are critical effector cells of the innate immune system.Under physiological conditions, entry of neutrophils into the brain is blocked by the blood-brain barrier; however, TBI-induced blood-brain barrier breakdown facilitates infiltration of neutrophils into the cerebral parenchyma (Liu et al., 2018).The ensuing neutrophil infiltration plays important roles in tissue repair and the elimination of cellular debris (Wang, 2018; Peiseler and Kubes, 2019; Alam et al., 2020).Unfortunately, aberrant neutrophil activation contributes to secondary brain injury and exacerbates neurological dysfunction after TBI (Jassam et al., 2017).Following TBI, infiltrated neutrophils upregulated N-acylethanolamine acid amidase, which in turn hydrolyzed palmitoylethanolamide and attenuated palmitoylethanolamide/peroxisome proliferator activated receptor α-mediated blood-brain barrier protection (Li et al., 2021).In addition, neutrophils caused tissue damage through the formation of NETs, which are extracellular web-like structures composed of DNA filaments decorated with histone and granule proteins,such as neutrophil elastase and myeloperoxidase (Papayannopoulos, 2018).TBI could activate the Toll-like receptor 4-peptidylarginine deiminase 4 cascade and promote the formation of NETs, which worsened cerebrovascular dysfunction and exacerbated neurological deficits (Vaibhav et al., 2020).Additionally, NETs induced by TBI in the paraventricular nucleus could evoke sympathetic hyperactivity through the LL37-Hippo/mammalian STE20-like protein kinases 1 pathway (Zhu et al., 2021).It is well established that neuroinflammation is highly involved in secondary brain injury (Jassam et al.,2017), although the underlying mechanisms remain elusive.More recently,Liu et al.(2021) showed that neutrophils trigger inflammatory responses and detrimental proinflammation-associated neurodegeneration following TBI through the Toll-like receptor 4 pathway, which could be mitigated by treatment with recombinant annexin A2.In the present work, we found that infiltrated neutrophils upregulated proinflammatory IL-17A and activated the NF-κB pathway, which implied that neutrophils might contribute to neuroinflammation-related secondary brain injury through the IL-17A cascade.
IL-17A is a pivotal player in various inflammatory diseases (McGeachy et al.,2019).In central nervous system diseases, growing evidence suggests that IL-17A is involved in the pathogenesis of secondary brain damage, such as ischemic stroke and TBI, in addition to multiple sclerosis and experimental autoimmune encephalomyelitis, which have been studied intensively (Li et al.,2017; Milovanovic et al., 2020; Zhang et al., 2021).Recent evidence showed that plasma IL-17A levels were significantly upregulated in patients with mild TBI from admission to follow-up at 3 months and remained elevated for up to 1 year, which hinted that IL-17A played a vital role throughout the clinical course of TBI-induced secondary injury (Chaban et al., 2020).Similarly, Li et al.(2017) demonstrated that IL-17A is increased in both the serum and central nervous system tissues in a rat model of TBI.Furthermore, IL-17A was found to promote neuronal apoptosis and compromise neural function through the IL-23/IL-17–mediated mitochondrial Bcl-2/Bax/caspase-3 signaling pathway.In line with these earlier studies, we observed IL-17A to be upregulated in the subacute phase of TBI, which was consistent with increased neuroinflammation.However, the cellular source of IL-17A identified in the current study differs from that described in previous studies.Here we found that neutrophils were the main source of IL-17A, whereas earlier studies have reported IL-17A to be largely secreted by T helper 17 cells.This apparent discrepancy may be attributable to differences in the animal model species,injury severity, and impact methods, or may indicate that IL-17A plays a multifaceted role in the pathophysiology of TBI.However, regarding the explicit functions of neutrophil-derived IL-17A in neuroinflammation after TBI, the present study had some limitations.First, the dynamic changes and detailed functions of neutrophil-derived IL-17A after TBI need to be further investigated.Most importantly, the mechanism of IL-17A–mediated cellularcommunication between infiltrated neutrophils and cerebral parenchymal cells remains to be elucidated.Finally, the sample size used in the present study was relatively small.
Of note, it has been shown that IL-17A could activate microglia and astrocytes, thereby exacerbating neuroinflammation and neurodegeneration(Ma et al., 2010; Liu et al., 2019; Chen et al., 2020; Sasaki et al., 2020).IL-17A signals through a heterodimeric complex composed of IL-17RA and IL-17RC, both of which are expressed in microglia and astrocyte.Upon interaction with its receptor, IL-17A canonically activates NF-κB–mediated proinflammatory responses (Chen et al., 2020).IL-17A could induce microglial neuroinflammation and promote microglial autophagy in intracerebral hemorrhage (Yu et al., 2016; Shi et al., 2018).Under inflammatory conditions, IL-17A triggered microglial activation and cognitive impairment in sepsis-associated encephalopathy and lipopolysaccharide-induced neuroinflammation (Sun et al., 2015; Ye et al., 2019).Intriguingly, cumulative mild stress could enhance IL-17A–mediated microglial activation in the hippocampus, amygdala, and prefrontal cortex, causing long-lasting anxietyand depression-like behavior (Kim et al., 2021).Based on the above findings,further investigation is needed to obtain a better understanding of the role of IL-17A–mediated crosstalk between infiltrated neutrophils and glia in neuroinflammation-induced injury.
In conclusion, neuroinflammation is believed to contribute to TBI-induced secondary brain injury; however, the underlying mechanism remained elusive.The overarching goal of this study was to determine the underlying factors that contributed to neutrophil-mediated neuroinflammation.To this end, histological and molecular biology analysis, coupled with nextgeneration sequencing, were used to characterize changes that occur in the brain after TBI.The results indicated that both astrocytes and microglia were highly activated 7 dpi.Moreover, IL-17A expression was upregulated, and IL-17A–associated pathways were significantly enriched.More interestingly,we found that IL-17A largely colocalized with activated neutrophils rather than microglia, astrocytes, or neurons, suggesting that neutrophils promote neuroinflammation via IL-17A cascades, thereby exacerbating the likelihood of inflammatory injury.Targeting neutrophil-derived IL-17A may be an innovative strategy to combat TBI-associated neuroinflammation.
Author contributions:
Study conception and design: XJX, BYL; animal experiment: XJX, FN; MRI examination: XJX, JQD; library construction, data analysis and immunofluorescence staining: XJX, QQG, MSY, YZ; RNA collection,qRT-PCR and Nissl staining: BZ, HL; manuscript draft and revision: XJX, BYL.All the authors approved the final version of the manuscript.
Conflicts of interest:
All authors in this manuscript have declared no conflict of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional file:
TBI induces abnormal gene expression in the ipsilateral cortex.
- 中国神经再生研究(英文版)的其它文章
- Patient-specific monocyte-derived microglia as a screening tool for neurodegenerative diseases
- Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases
- Inflammation in diabetic retinopathy: possible roles in pathogenesis and potential implications for therapy
- Targeting the nitric oxide/cGMP signaling pathway to treat chronic pain
- Neurosteroids as stress modulators and neurotherapeutics: lessons from the retina
- Myelinosome organelles in pathological retinas:ubiquitous presence and dual role in ocular proteostasis maintenance