
Alterations in gut microbiota are related to metabolite profiles in spinal cord injury
2022-09-27 09:45JianNingKangZhengFangSunXinYuLiXiaoDiZhangZhengXinJinCeZhangYingZhangHuiYunWangNaNaHuangJianHaoJiangBinNing
中国神经再生研究(英文版) 2023年5期
Jian-Ning Kang, Zheng-Fang Sun, Xin-Yu Li, Xiao-Di Zhang, Zheng-Xin Jin, Ce Zhang, Ying Zhang, Hui-Yun Wang,Na-Na Huang, Jian-Hao Jiang, , Bin Ning, ,
Abstract Studies have shown that gut microbiota metabolites can enter the central nervous system via the blood-spinal cord barrier and cause neuroinflammation,thus constituting secondary injury after spinal cord injury.To investigate the correlation between gut microbiota and metabolites and the possible mechanism underlying the effects of gut microbiota on secondary injury after spinal cord injury, in this study, we established mouse models of T8–T10 traumatic spinal cord injury.We used 16S rRNA gene amplicon sequencing and metabolomics to reveal the changes in gut microbiota and metabolites in fecal samples from the mouse model.Results showed a severe gut microbiota disturbance after spinal cord injury, which included marked increases in pro-inflammatory bacteria,such as Shigella, Bacteroides, Rikenella, Staphylococcus, and Mucispirillum and decreases in anti-inflammatory bacteria, such as Lactobacillus, Allobaculum,and Sutterella.Meanwhile, we identified 27 metabolites that decreased and 320 metabolites that increased in the injured spinal cord.Combined with pathway enrichment analysis, five markedly differential amino acids (L-leucine, L-methionine, L-phenylalanine, L-isoleucine and L-valine) were screened out, which play a pivotal role in activating oxidative stress and inflammatory responses following spinal cord injury.Integrated correlation analysis indicated that the alteration of gut microbiota was related to the differences in amino acids, which suggests that disturbances in gut microbiota might participate in the secondary injury through the accumulation of partial metabolites that activate oxidative stress and inflammatory responses.Findings from this study provide a new theoretical basis for improving the secondary injury after spinal cord injury through fecal microbial transplantation.
Key Words: 16S rRNA gene amplicon sequencing; amino acid metabolism; dysbacteriosis; gut microbiota; inflammation; metabolic disturbance; metabolites;metabolomics; secondary injury; spinal cord injury
Introduction
Spinal cord injury (SCI) often causes permanent paralysis, sexual dysfunction,and bladder/bowel dysfunction (Jogia and Ruitenberg, 2020; Kigerl et al.,2020).The overwhelming inflammatory response in the early stage of injury,combined with progressive spinal cord swelling and damage, results in dense glial and fibrous scars, leaving patients with sensory and motor dysfunction below the level of injury in the chronic stage (Hellenbrand et al., 2021).
Commensal gut microbiota play a crucial role in gut physiology and the regulation of host immune and endocrine systems, and their activity is closely related to the occurrence and development of a variety of host diseases(Round and Mazmanian, 2009; Clemente et al., 2012; Simrén et al., 2013;Sánchez et al., 2017; Zmora et al., 2019).The bidirectional communication between the gut microbial system and the host central nervous system (CNS)is achieved through the microbe-gut-brain axis (MGBA), involving immune,endocrine, metabolic, and neural pathways (Rhee et al., 2009; Cryan and Dinan, 2012; Forsythe and Kunze, 2013; Janssen and Kersten, 2015; Mayer et al., 2015; Fung et al., 2017; Baizabal-Carvallo, 2021).Recent findings demonstrate that the MGBA is also involved in the pathogenesis of a variety of CNS diseases, including Alzheimer’s disease (AD) (Wang et al., 2019),Parkinson’s disease (Scheperjans et al., 2015; Sampson et al., 2016), multiple sclerosis (Kadowaki and Quintana, 2020), stroke (Benakis et al., 2016), anxiety and depression (Forsythe and Kunze, 2013), and autism (Dan et al., 2020).However, there are few studies on the relevance of gut microbiota to SCI(Zhang et al., 2018; Bazzocchi et al., 2021; Li et al., 2022).
Some metabolites produced by gut microbiota are transported into the CNS and play crucial roles in the MGBA (Agus et al., 2018; Dalile et al., 2019).These include neuroactive metabolites (such as short-chain fatty acids,branched-chain amino acids, and peptidoglycans) and neurotransmitters(such as γ-aminobutyric acid, 5-hydroxytryptamine, dopamine, acetylcholine)(Tillisch, 2014; Cryan et al., 2019).Transport into the CNS is accomplished by increasing the permeability of the blood-brain barrier and intestinal mucosa barrier (Kigerl et al., 2020).Altered composition of intestinal microbiota can lead to metabolic disorders, with some microbial metabolites entering the CNS through the blood-spinal cord barrier and causing neuroinflammation(Erny et al., 2015; Rothhammer et al., 2016; Wang et al., 2019; Jing et al.,2021a).Metabolites act as a bridge between gut microbiota and the CNS, andthus the MGBA might have a role in SCI pathology.In this study, we assessed the correlation between gut microbiota and spinal cord metabolites, and explored how gut microbiota can impact secondary injury in SCI.
Methods
Animals and surgery
SCI model mice have flaccid paralysis of the hind limbs, impaired urination,and require manual urination assistance.Male mice have a narrow and long urethra, making urination difficult.This increases the risk of urinary tract infection in male mice, which not only increases the model’s mortality rate,but also has an impact on the detection of intestinal flora.Following a review of the literature, female mice were chosen for general SCI modeling, intestinal flora-related SCI modeling, and metabolism-related SCI modeling (Li et al.,2020a; Jing et al., 2021b; Milich et al., 2021).Seventy-two specific-pathogenfree level C57BL/6J female mice (age 6–8 weeks; weight 18–22 g) were purchased from Jinan Pengyue Laboratory Animal Breeding Co., Ltd.(license No.SCXK [Lu] 2019 0003, Jinan, China).All animals were housed at Shandong First Medical University in a specific-pathogen-free animal room at 23 ± 1°C and under a 12-hour light/dark cycle, with free access to standard mouse food and adequate water.Animal procedures were approved by Central Hospital Affiliated with Shandong First Medical University Welfare and Ethics of Laboratory Animals Committee (approval No.JNCHIACUC-202114) on September 14, 2021.All experiments were designed and reported according to Animal Research: Reporting ofIn Vivo
Experiments (ARRIVE) guidelines(Percie du Sert et al., 2022).Mice were randomly divided into sham and SCI groups (n
= 36 per group)(Figure 1).Mice were anesthetized by intraperitoneal injection of 3%pentobarbital (30 mg/kg; Sigma-Aldrich, St.Louis, MO, USA) followed by a T8–T10 laminectomy to expose the spinal cord.A mouse model of traumatic SCI was established by striking the spinal cord at a depth of 2 mm and a speed of 1 m/s using a spinal cord impactor (68100, RWD, Shenzhen, China).Traumatic SCI was deemed successful upon observation of significantly visible injury sites, transient spasms of the hindlimbs and tail, and loss of postoperative motor and sensory function (Li et al., 2021).After suturing the incisions layer by layer, the mice were placed on a heating blanket for 2 hours.Mice received an artificial bladder squeeze three times a day until spontaneous urination resumed.The sham group underwent laminectomy, but not SCI.All other procedures were the same for the two groups.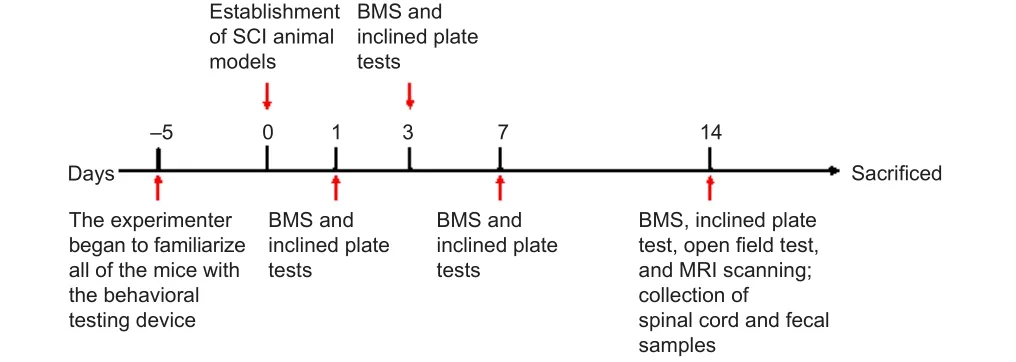
Figure 1|Timetable of animal experimental procedures.
Hematoxylin-eosin staining
Hematoxylin-eosin (HE) staining was performed to evaluate histopathological changes.Fourteen days post-injury (dpi), mice were anesthetized with 3% pentobarbital and cardiac perfusion was performed.T8–T10 spinal cord tissue from 5 mm above to 5 mm below the injury site was carefully collected and placed in 4% paraformaldehyde (Solarbio, Beijing, China) at 4°C overnight.These spinal cord tissue samples were then cut into 5-μm thick sections with a microtome (Leica (China), Shanghai, China).The prepared sections were placed into xylene and different concentrations of ethanol for deparaffinization.Hematoxylin staining (Solarbio) was then performed for 5 minutes, followed by washing in water and then washing in differentiation solution.Sections were sequentially dehydrated in 85% and 95% graded alcohol for 5 minutes each and stained in eosin staining solution for 5 minutes.Sections were then placed into each gradient of absolute ethanol for dehydration and mounted in neutral gum.Representative images were observed and captured using an electron microscope (Olympus, Tokyo,Japan).
Behavioral tests
Behavioral tests were performed at 0, 1, 3, 7, and 14 dpi.The recovery of hindlimb motor function in mice with SCI was evaluated using the Basso Mouse Scale (BMS) score, inclined plate test, and open field test.All experiments were performed with the participation of well-trained,experienced technicians who were unaware of the animal groupings.
BMS scores
The BMS score, which ranges from 0 (complete paralysis) to 9 (normal mobility), represents the state of hindlimb motor function recovery after SCI in mice (Basso et al., 2006; Zhou et al., 2020).Mice were initially placed in an open field every day for 5 days before surgery to become used to walking environment.All mice were evaluated by the same observers who were familiar with the scoring rules and blind to the experimental conditions.
Inclined plate test
The inclined plate test was used to assess an animal’s ability to maintain its position on a board raised in 5° increments.Mice were placed on the inclined plate with the longitudinal axis of the body parallel with that of the plate,and with the head on the elevated side.The maximum angle at which the mouse remained in this position for 5 seconds without slipping was recorded.Measurements were taken 5 times per animal, and the mean inclined plate angle was used as the index value.The angle of the inclined plate mirrors the weight-bearing capacity and recovery status of the animal’s hind limbs (Wells et al., 2003).
Open field test
At 14 dpi, mice performed the open field test with the Video Analysis System(Saeons, Jiangsu, China).The Video Analysis System comprises a 45 cm ×45 cm × 35 cm chamber and a small animal infrared autonomous testing system.Mice were placed in the corner of the chamber, and were allowed to move freely for 5 minutes.The system automatically calculates the mouse’s movement trajectory, distance walked, number of times they stood up, and the duration of standing.
T2-weighted magnetic resonance imaging
Two groups of mice were tested at 14 dpi using a small animal 9.4-T MRI scanner (Bruker, 9.4T Biospec; Bruker BioSpin, Germany) with a four-channel surface coil.Inhalation anesthesia was administered with 1.5% isoflurane(RWD, Shenzhen, China) using a small animal anesthesia set matched to MR before mice were placed on a specialized fixation system.The following parameters were used in the sequence protocol: T2-weighted; 320 × 320 matrix; slice thickness = 0.3 mm; echo time/repetition time = 24/1200 ms; flip angle = 90°.The Bruker ParaVision 6.0 system (Bruker, Ettlingen, Germany)was used to obtain T2-weighted images in the sagittal plane.After that, the mice were placed on a heating pad to recover.
16S rRNA gene amplicon sequencing
At 14 dpi, fecal samples were collected in 2.0-mL sterile tubes, snap frozen in liquid nitrogen, and stored at –80°C for further analysis.The fecal samples were then placed in dry ice and sent to Shanghai Personal Biotechnology Co., Ltd.(Shanghai, China) for 16S rRNA Gene Amplicon Sequencing and Bioinformatics and Statistical Analysis.Microbial genomic DNA was extracted using the OMEGA Stool DNA Kit (D4015-02, Omega Bio-Tek, Norcross, GA,USA) according to the manufacturer’s instructions.The quality of extracted DNA was examined by agarose gel electrophoresis, and quantified using a NanoDrop NC2000 spectrophotometer (Thermo Fisher Scientific, Waltham,MA, USA).PCR amplification of the bacterial 16S rRNA genes V3–V4 region was performed using the forward primer 338F (5′-ACT CCT ACG GGA GGC AGC A-3′) and the reverse primer 806R (5′-GGA CTA CHV GGG TWT CTA AT-3′).After the individual quantification step, amplicons were pooled in equal amounts, and pair-end 2× 250 bp sequencing was performed based on the Illumina NovaSeq platform.Microbiome bioinformatics was performed on the QIIME2 platform of Shanghai Personal Biotechnology Co., Ltd.
α-Diversity and β-diversity were assessed with the QIIME2 platform (https://qiime2.org/) (Bolyen et al., 2019).PICRUSt 2 (https://github.com/picrust/picrust2) was applied to predict and analyze the microbial diversity function(Langille et al., 2013).The relative abundance of secondary functional pathways was analyzed based on the MetaCyc database (https://metacyc.org/) (Karp et al., 2019).
Widely targeted metabolomics
The spinal cord tissue 1.0 cm around the injury site was collected in 2.0-mL sterile tubes, preserved in liquid nitrogen, and stored at –80°C before use.At the time of detection, the samples were removed from the –80°C refrigerator and thawed on ice.Each sample that weights 20 mg was ground thoroughly in liquid nitrogen.Then, the samples were mixed with 400 μL of 70%methanol/water internal standard extractant, shaken (100 ×g
) for 5 minutes,iced for 15 minutes, and centrifuged (16,260 ×g
4°C) for 10 minutes.The supernatant was stored at –20°C for 30 minutes and centrifuged (16,260 ×g
,4°C) for 3 minutes before being collected for analysis.Metabolite detection,identification, and quantification experiments on the collected supernatant were performed in a liquid chromatography-electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) system provided by Wuhan MetWare Biotechnology Co., Ltd.(Wuhan, China).Mass spectrometry data analysis was provided by Wuhan MetWare Biotechnology Co., Ltd., and all analytical data were processed using Analyst 1.63 software (Sciex, Framingham, MA,USA) based on the self-built database MetWare (MWDB, Wuhan MetWare Biotechnology Co., Ltd.).Based on a Principal Component Analysis separationoptimization model, orthogonal partial least squares discrimination analysis containing score plots and permutation plots was generated.Variable importance in projection (VIP) values were extracted from the results of the orthogonal partial least squares discrimination analysis.The above process was accomplished by the MetaboAnalystR package for R (https://www.metaboanalyst.ca/).Targeted metabolomics
At 14 dpi, T8–T10 spinal cord tissue, including 1.0-cm around the injury site, was collected in 2.0-mL sterile tubes, preserved in liquid nitrogen, and stored at –80°C.After being thawed and smashed on ice, the samples were mixed with 500 μL of 70% methanol/water (precooled to –20°C in advance).Samples were shaken at 280 ×g
for 5 minutes, and centrifuged for 10 minutes at 16,260 ×g
at 4°C.The supernatant was transferred to a new centrifugetube, placed in a –20°C refrigerator for 30 minutes, and centrifuged at 16,260×g
for 10 minutes at 4°C before being transferred to a protein precipitation plate for further LC-MS analysis.Metabolite detection, identification, and quantification experiments on the collected supernatant were performed in the LC-ESI-MS/MS system provided by Wuhan MetWare Biotechnology Co.,Ltd.Qualitative analysis of mass spectrometry data was performed using the MWDB (MetWare Database).The mass spectrometry data were processed using Analyst 1.6.3 software (Sciex, Framingham, MA, USA).The mass spectrometry peak-intensity data were collected corresponding to different concentrations of standard solutions.The integrated peak-area ratio of all detected samples was calculated by substituting the linear equation of the standard curve and then substituting the calculation formula to finally obtain the data for the substance in the actual sample.Statistical analysis
The sample size was determined based on the minimum number of samples required by the sequencing company.Statistical analyses were performed using SPSS 19.0 software package (IBM Corp., Armonk, NY, USA).Data are presented as mean ± standard deviation (SD).BMS scores and inclined plate test scores were analyzed with a two-way analysis of variance (ANOVA)followed by Bonferronipost hoc
correction.The α-diversity was analyzed by Kruskal-Wallis test and β-diversity was assessed with an Adonis and Anosim analysis.Relative bacterial content and metabolite content were compared with a Student’st
-test.Spearman correlation analysis was used to determine correlation coefficients between gut microbial changes and major differential metabolites in SCI.Statistical significance was set atP
< 0.05.Results
A persistent inflammatory response occurs after SCI
To confirm the successful establishment of the mouse model of SCI using an SCI impactor, motor function was assessed by behavioral tests (BMS score,inclined plate test, and open field test) at 1, 3, 7, and 14 dpi (Figure 2A–D).Hindlimb paralysis immediately developed in the SCI group, but did not in the sham group.Mouse spinal cords were collected for sagittal sectioning at 14 dpi, and HE staining (Figure 2E) was used to observe histopathologic changes.In the sham group, the spinal cord structures remained intact without hemorrhage, necrosis, or inflammatory cell infiltration.In contrast, the SCI group was characterized by rupture of spinal cord structures, accompanied by hemorrhage, inflammatory cell infiltration, and neuronal loss.T2-weighted MR images of sagittal slices of mouse spinal cord in each group are shown at 14 dpi in Figure 2F.A disruption of the continuity of the spinal cord structures can be seen in the MR images.There is a significant hypointense area at the lesion, indicating the presence of stale hemorrhage, and scattered hyperintense areas surrounding the lesion, indicating edema formation.Therefore, a persistent inflammatory response existed after SCI.
Gut microbial composition is different between the sham and SCI groups
To investigate whether the gut microbiota were different between the two groups fecal samples were collected at 14 dpi and 16S rRNA gene amplicon sequencing was used to compare the fecal microbiota composition between the sham (n
= 22) and SCI groups (n
= 22).The α-diversity indexes were used to compare microbial diversity and richness (Patrick et al., 2020).As shown in Figure 3A, compared with the sham group, the Chao1 and observed species indexes, which represent species richness, were significantly lower than those in the SCI group, and the Shannon and Simpson indexes, which represent species diversity, also trended lower.The Pielou’s species evenness index was also lower in the SCI group than in the sham group.These findings suggest that SCI seems to have reduced microbiota diversity.Based on Bray-Curtis, β-diversity analysis was carried out to determine whether the observed difference in gut microbial composition was caused by SCI.The results of principal-coordinate analysis and nonmetric multidimensional scaling demonstrated that the gut microbiota from fecal samples differed between the sham and SCI groups (Figure 3B and C).Similarly, Adonis and Anosim tests also revealed that the community structure and composition of gut microbiota differed significantly between groups (Figure 3D and E), indicating that SCI caused gut microbiota dysbiosis.Furthermore, hierarchical clustering analysis was performed using an unweighted pair-group method with arithmetic means.As shown in Figure 3F, the sham and SCI samples formed a separate group.
Based on the results of species annotation, the top 10 species were selected in each group at the phylum and genus level (Figure 4A and B).Bacteroidetes,Firmicutes, and Proteobacteria dominated the gut microbiota at the phylum level.Specifically, the intestinal contents of the SCI mice contained a higher abundance of Firmicutes and Proteobacteria but a lower abundance of Bacteroidetes.The Firmicutes/Bacteroidetes (F/B) ratio reflects the degree of dysbiosis of the gut microbiota (Feng et al., 2019), and was significantly higher in the SCI group than in the sham group (Figure 4C and D).At the genus level,Lactobacillus
,Shigella
,Prevotella
,Bacteroides
, andAllobaculum
were the major identified genera.Comparing the two groups revealed that the SCI group exhibited a lower relative abundance ofLactobacillus
,Allobaculum
, andSutterella
, and a higher relative abundance ofShigella
andBacteroides
.
Figure 2|Motor and histopathologic changes in the mouse model of SCI.
To study the common and unique species among the different groups of mice, we used a Venn diagram for community analysis (Figure 5A).The Venn diagram was created using the amplicon sequence variants (ASV) abundance table, which counted the number of members of each set (the number of ASV unique to each group and common between groups) based on their presence or absence between groups.Figure 5A shows that 5365 ASVs were common to both groups, accounting for 18.92% of those in the sham group and 33.50%of those the SCI group.The Sham group contained more unique ASVs than the SCI group, which is consistent with the higher Chao1 index seen in Figure 3A.Linear discriminant analysis (LDA) effect size was determined to find the bacterial taxa that differed significantly between groups at all taxonomic levels (P
< 0.05, LDA > 4; Figure 5B and Additional Figure 1).The analysis revealed marked differences between the microflora of SCI and sham mice,characterized by higher levels of Firmicutes and lower levels of Bacteroidetes in the SCI mice.Bacteroides
,Rikenella
, andShigella
dominated in the SCI group, whileLactobacillus
,Allobaculum
, andSutterella
were more prevalent in the sham group.These different characteristic bacteria might affect host health in a variety of ways.A genus-level heatmap further demonstrates the effect of SCI on the organization of gut microbiota (Figure 5C).As in the previous analysis,amounts ofStaphylococcus
,Corynebacterium
,Shigella
,Odoribacter
,Rikenella
, andBacteroides
were significantly higher in the SCI group than in the Sham group, while amounts ofAllobaculum
,Sutterella
, andLactobacillus
were lower.Random forest analysis was applied to screen out the marker species that have an important influence on the differences between groups(Dicker et al., 2021).This analysis can deeply probe the complex nonlinear interdependence between variables, and the algorithm can classify microbial community samples effectively, robustly, and accurately.As shown in Figure 5D, abundance ofAllobaculum
,Phascolarctobacterium
,Bifidobacterium
,andSutterella
was significantly high in the sham group, indicating its representative strains.Simultaneously, high-contentStaphylococcus
,Bacteroides
,Odoribacter
,Mucispirillum
, andRikenella
became the dominant strains of the SCI group.These results are generally consistent with the heatmap and LDA effect size analysis, indicating that the gut microbial community was dysregulated, a phenomenon that can occur following SCI and be accompanied by the enrichment of proinflammatory bacteria, such asShigella
,Bacteroides
, Rikenella
,Staphylococcus
, andMucispirillum
(Wallace et al., 2011; Cattaneo et al., 2017; Cryan et al., 2020; Li et al., 2020b; Shintouo et al., 2020; Guo et al., 2021; Kim et al., 2021; You et al., 2022) and the loss of anti-inflammatory bacteria, such asLactobacillus
,Allobaculum
, andSutterella
(Morgan et al., 2015; Jangi et al., 2016; Butera et al., 2020; Pujo et al., 2021).We predicted microbiota function using Picrust2 software to investigate whether differences in the composition of gut microbiota were associated with functional changes.The function of gut microbiota in terms of their hosts is primarily biosynthesis, especially amino acids, nucleotides, vitamins,fatty acids, and lipids (Additional Figure 2).Based on our findings in the gut microbiome, metabolomics was further tested to confirm whether local metabolites were altered after SCI.
Figure 3 | α-Diversity and β-diversity based on ASV level in gut microbiota of the sham and SCI groups.

Figure 4 | Shift in the composition of gut microbiota species following SCI.

Figure 5|Importance of marker species between the sham and SCI groups was plotted on the graph.
Intestinal bacteria-derived metabolites differed significantly after SCI
Intestinal bacteria-derived metabolites affect host pathophysiological processes through multiple pathways (Olson et al., 2018; Cryan et al., 2020).Widely targeted metabolomics was used to determine spinal cord tissue metabolic profiles and to analyze the relationship between gut microbiota and these metabolites.An excellent separation between the sham group and the SCI group was revealed by orthogonal partial least squares discrimination analysis, which indicated that SCI led to severe metabolic dysfunction (Figure 6A).Furthermore, 200 permutation tests (R
= 0.603,R
= 0.996,Q
= 0.95 andP
< 0.005) showed that the orthogonal partial least squares discrimination analysis model had reliable predictive power (Figure 6B).The S-plot of the results of the analysis shows the differential expression of metabolites with VIP values greater than 1 or less than 1 between the two groups (Figure 6C).The volcano diagram was used to screen out differential components from all the detected metabolites and was based on VIP values and foldchange variations (VIP > 1.0, fold change > 2 / < 0.5) after SCI (Figure 6D).The diagram indicated that in the SCI group, levels of 27 metabolites were significantly lower and levels of 320 metabolites were significantly higher.Subsequently, a heatmap was used to classify and display these metabolites(Additional Figure 3).To identify potential biomarkers, the top 10 metabolites that were higher and the top 10 that were lower were plotted in bar graphs(Additional Figure 4).The relative content of differential metabolites with VIP values in the top 50 is shown by violin diagram (Additional Figure 5).These metabolites whose levels differed significantly depending on group included amino acids and their metabolites, glyceryl phosphatide, fatty acyls,and organic acids and their derivatives.The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was applied in an enrichment analysis of differential metabolites (Figure 6E).Among all the enriched pathways, amino acid metabolism-related pathways were the most abundant.Combined with the KEGG pathway enrichment results, five metabolites, including L-leucine,L-methionine, L-phenylalanine, L-isoleucine and L-valine, were selected as markers that might cause some effects at the site of injury.The excessive accumulation of these five amino acids might induce oxidative stress and inflammatory responses, and thus participate in secondary injury after SCI.Correlation analysis between gut microbiota and local metabolites
The potential correlation between the altered gut microbiota and local metabolites after SCI was investigated in depth by calculating the Spearman’s rank correlation coefficient.Correlation analysis between the top 50 differential metabolites and the top 20 differential microbiota communities at the genus level was performed and detected significant correlations between some bacteria and multiple different metabolites (|r
| > 0.6 andP
< 0.05;Figure 7A).These includedLactobacillus
,Shigella
,Mucispirillum
,Sutterella
,andAllobaculum
.These microbiota might impact metabolic processes after SCI, correlating with the accumulation of certain metabolites.Some amino acids, such as L-leucine, L-methionine, L-phenylalanine L-isoleucine,and L-valine, increased significantly after SCI and correlated positively with Shigella and Mucispirillum, but negatively with Lactobacillus, Sutterella, and Allobaculum (Figure 7B).Targeted metabolomics was performed to validate the content of target metabolites after SCI (Figure 7C).In summary, these results suggest that the changes in gut microbiota correlated with the certain metabolites, which might in turn aggravate the inflammatory response to SCI.However, whether these metabolites are directly influenced by altered gut microbes warrants further investigation.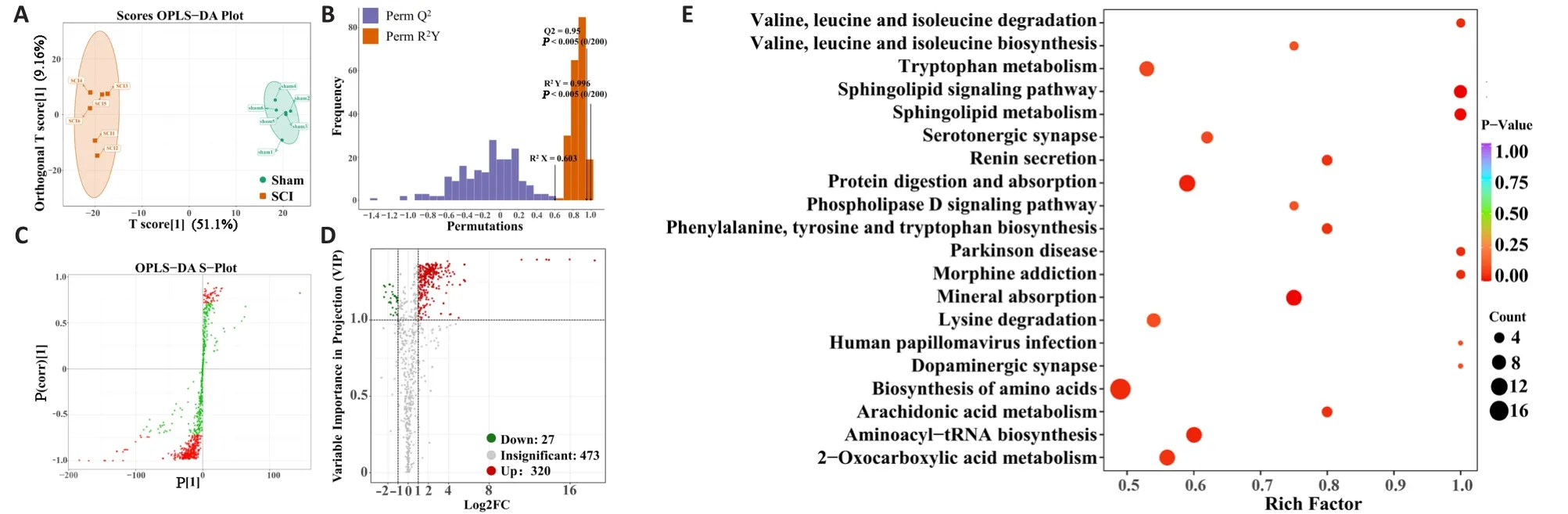
Figure 6 | Screening of differential metabolites after spinal cord injury.
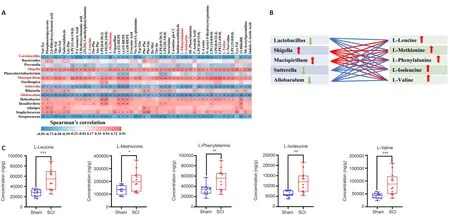
Figure 7 | Correlation analysis between the relative abundances of the microbial genera and the spinal cord metabolites after SCI.
Discussion
Studies of SCI have mostly focused on changes in DNA, RNA, and protein.However, these studies have not brought a full understanding of the systemic changes after SCI.An increasing number of studies have shown that the combined detection of microbiomes and their related metabolites may help us understand systemic changes that occur with diseases, as well as explore the possible mechanisms of their progression (Franzosa et al., 2019; Dan et al.,2020).In this study, 16S rRNA gene amplicon sequencing and metabolomics were used to reveal differences in gut microbiota and spinal cord metabolites between mice with and without SCI.We then investigated the correlation between these two factors.Our findings suggest that both microbial composition in the gut and metabolites in the spinal cord were altered in the mouse model of SCI.Simultaneously, correlation analysis showed that important metabolites that were differentially found in the SCI group were closely associated with the top 20 gut microbiota that were present after SCI.Our results thus reveal that gut microbiota might be involved in the local inflammatory response within the area of SCI by causing the accumulation of certain amino acids.These results are supported by Kigerl et al.(2016), which reported that the introduction and/or augmentation of beneficial microbial communities may be warranted to modify the inflammatory response in favor of improved neurological outcomes.In our study, we focused on the potential correlation between the altered composition of gut microbiota and the altered local metabolites in the spinal cord after SCI.Known correlations can provide a future direction for exploring the systemic changes after SCI.
In the current study, gut microbiome analysis showed that microbial diversity and richness decreased after SCI, which could be caused by a combination of symptoms, such as bladder/bowel dysfunction, dietary structure changes,and motor dysfunction (Kigerl et al., 2018).However, the F/B ratio increased,which indicates an imbalance in health status and dysbiosis of gut microbiota(Grigor’eva, 2020; Stojanov et al., 2020).In previous studies, an increased F/B ratio has been associated with several neurological disorders, such as neuropathic pain, cognitive impairment, and stroke (Park et al., 2020; Yang et al., 2020).Previous findings also indicate that disturbances of gut microbiota might contribute to the chronic inflammation stage of secondary injury (Du et al., 2021).However, these are only one-sided indicators for assessing the effects of the entire microbiome on SCI and more indicators need to be analyzed together.The data obtained from 16S rRNA gene-amplicon sequencing demonstrate that the relative abundance ofShigella
,Bacteroides
,Rikenella
,Staphylococcu
s, andMucispirillum
increased in the SCI group, while that ofLactobacillus
,Allobaculum
, andSutterella
decreased.Cross-sectional studies have found that Shigella enrichment can trigger or exacerbate neurodegeneration in patients with AD (Cattaneo et al., 2017; Cryan et al.,2020).Guo et al.(2021) found that Bacteroides disrupted the intestinal barrier and aggravated host systemic inflammation when proliferation was excessive.Another study has also shown that proliferation of Bacteroides might be causally related to systemic inflammation, this case being in a model of unruptured intracranial aneurysms (Li et al., 2020b).In a recent study of AD, researchers found that Rikenella might lead to microglial activation in the mouse hippocampus, promote proinflammatory cytokine expression(such as tumor necrosis factor-α and interleukin-1β), significantly reduce hippocampal neurons in mice, and cause learning and memory impairment in mice (Kim et al., 2021).As a core member of mouse gut microbiota,Mucispirillum has been shown to be positively correlated with monocyte chemotactic protein-1 (Shintouo et al., 2020).You et al.(2022) also found that specific bacterial taxa such as Staphylococci might be associated with lower bile acid metabolism levels after traumatic brain injury, which leads to intestinal inflammation.However, Lactobacillus and Allobaculum were found to exert key anti-inflammatory properties in various diseases such as stroke, colitis, and arthritis (Lee et al., 2020; Pujo et al., 2021).Similarly, other studies have found that Sutterella abundance was negatively correlated with levels of inflammatory factors (Morgan et al., 2015; Jangi et al., 2016; Butera et al., 2020).Interestingly, in our study, a similar phenomenon was found:proinflammatory bacteria proliferated and anti-inflammatory bacteria were inhibited.This supports a correlation between altered gut microbes and the inflammatory response around the lesion site.Serum or local metabolites are thought to be closely related to the gut microbiota after SCI.Although previous studies focused on serum metabolites after SCI, this study has shifted focus to local metabolites of the injured spinal cord, which represent a real abnormal metabolism after SCI and provide new biomarkers and therapeutic strategies for treating SCI.The distinguishing metabolites between groupsin our study were primarily amino acids and their metabolites, glyceryl phosphatide, fatty acyls, and organic acids and their derivatives.Five amino acids (L-leucine, L-methionine, L-phenylalanine,L-isoleucine, L-valine) were found to be significantly enriched after SCI, and we propose them to be important biomarkers of SCI.Evidence that these five amino acids are involved in neuroinflammation has accumulated over years of research.Previous studies in multiple sclerosis have found that methionine restriction limits the proliferation of T helper 17 cells through cellular epigenetic reprogramming, which in turn reduces the morbidity and severity of experimental autoimmune encephalomyelitis (Roy et al., 2020).The critical role of amino acid metabolism in AD has also been recently demonstrated.The progression of AD is often accompanied by peripheral accumulation of phenylalanine and isoleucine, which induce the activation of M1 microglia by stimulating the differentiation and proliferation of T helper 1 cells, leading to Alzheimer’s-related neuroinflammation (Wang et al., 2019).Furthermore,elevated content of branched-chain amino acids (leucine, isoleucine and valine) promoted the activation of circulating peripheral blood mononuclear cells and contributed to inflammation and oxidative stress through reactive oxygen species production and nuclear factor kappa-B pathway (Zhenyukh et al., 2017).To further validate our experimental results, targeted metabolomics was performed to quantify these five target metabolites, and the results demonstrated that their amounts significantly increased after SCI.Unlike previous metabolomics studies of SCI that focused on short-chain fatty acids, our study found that amino acid metabolism may be closely related to oxidative stress and inflammation after SCI.Studying the abnormal amino acid metabolism may provide new insights into the pathogenesis of secondary injury after SCI.
Our study still has some limitations.Although, the results we provided demonstrate that changes in gut microbiota might be related to SCI, further fecal bacterial transplantation experiments are needed in the future to verify the causal relationship.The five characteristic amino acids identified in this experiment need to be further validated in the future to determine their relationship with neuroinflammatory responses.When combining the 16S rRNA gene-amplicon sequencing with metabolomics, a reasonable deduction is that disturbances in gut microbiota might participate in the secondary injury via accumulated partial amino acid metabolites, which trigger oxidative stress and inflammatory responses.This study may provide a new medical theoretical basis for the correlation between gut microbiota and related metabolites in SCI.
In conclusion, correlation analysis revealed a significant linear relationship between gut microbiota and characteristic metabolites.The presence of characteristic amino acids, such as L-leucine, L-methionine, L-phenylalanine,L-isoleucine, and L-valine, which were more abundant after SCI, were correlated significantly with the amount of Shigella, Mucispirillum,Lactobacillus, Sutterella, and Allobaculum.This suggests that disturbance in gut microbiota might lead to the accumulation of certain amino acids that participate in inflammatory responses surrounding the lesion.However,further investigation is needed to determine whether the characteristic amino acids are direct metabolites of the altered gut microbes and which microbe play central roles.
Acknowledgments:
The authors would like to acknowledge the following institutions: Central Hospital Affiliated to Shandong First Medical University’s Research Center of Basic Medicine, Research Center of Translational Medicine,and Laboratory Animal Center offered experimental support.The image of graphic abstract was authorized and created in BioRender.com.
Author contributions:
Study design: BN, JNK, JHJ; experiment implementation and data collection: JNK, ZFS, XYL, XDZ, ZXJ, CZ, YZ, HYW, NNH; data analysis:JNK, JHJ, ZFS, XYL, XDZ, ZXJ, CZ, YZ, HYW, NNH; manuscript draft: JNK; study supervision: BN.All authors approved the final version of the manuscript.
Conflicts of interest:
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:
Willemijn Faber, Heliomare Rehabilitation Centre,Netherlands; Gabriele Bazzocchi, Montecatone Rehabilitation Institute, Italy.
Additional files:
LDA chart scores as obtained by LDA analysis.
Relative abundance of metabolic pathways (MetaCyc database) at the secondary classification level of gut microbiota.
Heat map showing the relative content of different metabolites for all experimental groups.
Bar charts showing the top 10 increased (red) and decreased (green) metabolites.
The top 50 differential metabolites selected by violin plot analysis between Sham and SCI groups.
- 中国神经再生研究(英文版)的其它文章
- Patient-specific monocyte-derived microglia as a screening tool for neurodegenerative diseases
- Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases
- Inflammation in diabetic retinopathy: possible roles in pathogenesis and potential implications for therapy
- Targeting the nitric oxide/cGMP signaling pathway to treat chronic pain
- Neurosteroids as stress modulators and neurotherapeutics: lessons from the retina
- Myelinosome organelles in pathological retinas:ubiquitous presence and dual role in ocular proteostasis maintenance