
Mechanism of piR-1245/PIWI-like protein-2 regulating Janus kinase-2/signal transducer and activator of transcription-3/vascular endothelial growth factor signaling pathway in retinal neovascularization
2022-09-27 09:45YongYuLiKunXiaYuDiQingZhuNieXiaoLongChen
中国神经再生研究(英文版) 2023年5期
Yong Yu, Li-Kun Xia, Yu Di, Qing-Zhu Nie, Xiao-Long Chen
Abstract Inhibiting retinal neovascularization is the optimal strategy for the treatment of retina-related diseases, but there is currently no effective treatment for retinal neovascularization.P-element-induced wimpy testis (PIWI)-interacting RNA (piRNA) is a type of small non-coding RNA implicated in a variety of diseases.In this study, we found that the expression of piR-1245 and the interacting protein PIWIL2 were remarkably increased in human retinal endothelial cells cultured in a hypoxic environment, and cell apoptosis, migration, tube formation and proliferation were remarkably enhanced in these cells.Knocking down piR-1245 inhibited the above phenomena.After intervention by a p-JAK2 activator, piR-1245 decreased the expression of hypoxia inducible factor-1α and vascular endothelial growth factor through the JAK2/STAT3 pathway.For in vivo analysis, 7-day-old newborn mice were raised in 75 ± 2% hyperoxia for 5 days and then piR-1245 in the retina was knocked down.In these mice, the number of newly formed vessels in the retina was decreased, the expressions of inflammationrelated proteins were reduced, the number of apoptotic cells in the retina was decreased, the JAK2/STAT3 pathway was inhibited, and the expressions of hypoxia inducible factor-1α and vascular endothelial growth factor were decreased.Injection of the JAK2 inhibitor JAK2/TYK2-IN-1 into the vitreous cavity inhibited retinal neovascularization in mice and reduced expression of hypoxia inducible factor-1α and vascular endothelial growth factor.These findings suggest that piR-1245 activates the JAK2/STAT3 pathway, regulates the expression of hypoxia inducible factor-1α and vascular endothelial growth factor, and promotes retinal neovascularization.Therefore, piR-1245 may be a new therapeutic target for retinal neovascularization.
Key Words: angiogenesis; human retinal endothelial cells; hypoxia inducible factor-1α; hypoxia; interleukin-1β; migration; non-coding RNA; oxygen-induced injury; PIWI-interacting RNA; retinopathy
Introduction
Retinal neovascularization (RNV) is a pathological feature of various retinal diseases and a serious ocular condition that can cause blindness.The prevalence of RNV is gradually increasing each year (Manian et al., 2021;Zhang et al., 2021a).However, the mechanisms underlying its pathogenesis are not well understood.At present, RNV inhibition is the main strategy for the treatment of related retinal diseases, with treatment methods such as retinal laser photocoagulation, intraocular injection of vascular endothelial growth factor (VEGF) inhibitors, and other approaches (Geng et al., 2018;Mitchell et al., 2021).These treatments can mitigate disease progression in a small number of patients by reducing the number of newly formed blood vessels.However, their widespread use has been limited by considerable adverse effects, including laser thermal damage to normal retinal blood vessels, drug-induced damage to choroidal vasculature, and the development of drug resistance (Geng et al., 2018; Mitchell et al., 2021).Therefore, better understanding of the molecular mechanisms underlying RNV is critical to identify novel therapeutic targets and develop alternative or adjuvant therapies without adverse effects for patients with RNV-related diseases.
Neovascularization (NV) is the formation of new blood vessels and capillary beds from existing blood vessels and it plays a crucial role in numerous physiological and pathological processes (Zhang et al., 2019).NV is extremely complex, requiring coordination among multiple cytokines and the activation of different cellular mechanisms (Elshaer and El-Remessy, 2018).The most important process in NV involves the activation of endothelial cells (ECs),which induces sprouting, proliferation, and degradation of the extracellular basement membrane, paving the way for migration, lumen formation, and finally vascular maturation and blood perfusion (Mather et al., 2021).Thetightly connected retinal microvascular ECs are the major cellular constituents making up the blood-retinal barrier (Lee et al., 2021).As such, hypoxiaassociated changes in their extracellular matrix as well as proliferation and migration stimulated by VEGF and other cytokines are crucial to the NV process (Hu et al., 2020).Recent studies have shown that piRNA is involved in the onset and development of NV in various tumors, which is suggestive of a role for piRNA in RNV disease progression (Tantawy et al., 2020; Zhou et al.,2020).The Janus kinase-2/signal transducers and activators of transcription-3(JAK2/STAT3) pathway, a major signaling cascade that regulates diverse cell functions, such as proliferation, differentiation, and cell transformation,promotes NV and the recruitment of inflammatory cells (Norvilas et al., 2021).JAK-STAT is abnormally expressed in a wide range of cancer tissues and cells(Xu et al., 2013; Wang et al., 2021).Studies have shown that STAT3-driven VEGF production can remarkably enhance the proliferation and migration of vascular ECs, while also participating in and accelerating vascular remodeling(Cheng et al., 2020; Lin et al., 2021).JAK2 and STAT3 are key upstream molecules of VEGF angiogenic signaling vital to the regulation of EC function,including angiogenesis (Melamed and Kalderon, 2020).
Studies have shown that non-coding RNAs play key functions in malignant tumor transformation and in vascular diseases (Mather et al., 2021; Thuillier and Behm-Ansmant, 2021).P-element-induced wimpy testis (PIWI)-interacting RNA (piRNA) is a small non-coding RNA implicated in multiple diseases.PIWI/piRNA complexes influence somatic cells and cancer tissue by regulating cell proliferation, apoptosis, cell cycle blockade, angiogenesis, invasion, and metastasis (Ipsaro et al., 2021; Zeng et al., 2021).PIWI also interacts with PIWI-like proteins (PIWILs), a family that includes PIWIL1, PIWIL2, PIWIL3 and PIWIL4.The piRNA-PIWI complex is normally found within the cytoplasm.Similar to the mechanism of siRNA, this complex can destroy target mRNAs translocated from the nucleus, silencing target gene expression (Sukthaworn et al., 2019; Yamashiro et al., 2020).Thus, exploring the functions of piRNA-coupled proteins will elucidate the biological functions of piRNAs.On the basis of research demonstrating that piRNAs play key roles in NV-related disease,we explored the potential role of piR-1245 in RNV.We established a human retinal EC (HREC) model in hypoxia conditions and the classic mouse model of oxygen-induced retinopathy (OIR).piR-1245 knockdown was performed to study the expression levels of retina-related indicators and its effect on retinal EC function.Elucidating the potential role of piR-1245 in RNV pathogenesis will provide a basis for subsequent research and the development of therapeutics.
Methods
Cell culture and reagents
HRECs (RRID: CVCL_B5WJ) were purchased from the National Collection of Authenticated Cell Cultures (Shanghai, China).The cell line was authenticated after identification and mycoplasma detection.HRECs were cultured in RPMI 1640 (HyClone, Shanghai, China) containing 10% fetal bovine serum (Gibco,New York, NY, USA) at 37°C and 5% CO.
For hypoxic culture, cells were cultured in medium containing 200 μM of the hypoxia mimetic CoCl(Sigma, St.Louis, MO, USA) for 48 hours (Wang et al.,2022).In some experiments, a p-JAK2 activator (Cat# 420139, Sigma) was used (10.6 μM) after 12 hours of hypoxic culture (Figure 1).
Figure 1| Schematic of treatment groups and cell experiments.
Cell transfection
Cells were transfected using the Lipofectamine 3000 reagent (Invitrogen,Waltham, MA, USA) following the manufacturer’s instructions.Cells at 70–90% confluence were used for transfection.Opti-MEM medium (Gibco)and Lipofectamine 3000 reagent (Invitrogen, Waltham, MA, USA) were mixed with 2.5 μg of sh-piR-1245 plasmid (Genchem Co., Shanghai, China) in cell culture medium.The sh-NC group was added with empty plasmid (Genchem,Shanghai, China).The plasmid was designed and synthesized by Genchem.
Scratch assay
A marker pen was first used to draw parallel straight lines spaced 0.5–1.0 cm apart on the back of 6-well plates.Cells were evenly inoculated into the cell culture plate.The next day, a pipette tip was used to make scratches in the cell monolayer perpendicular to the horizontal lines on the back of the plate.The plate was then gently washed three times with phosphate-buffered saline(PBS) to remove the detached cells, replenished with serum-free medium, and incubated at 37°C and 5% CO.At 0 and 48 hours after culture, the growth and migration of cells were observed using a fluorescence inverted microscope(ECLIPSE Ti, Nikon, Tokyo, Japan).The width of the scratch was measured scratch boundary of each group was measured as the distance of cell migration for statistical analysis.The migration distance of cells was analyzed by ImageJ 1.37C software (National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012).Wound healing rate was calculated as follows: migration (%) = (0 hour scratch width – 48 hours scratch width)/0 hour scratch width × 100 (Yao et al., 2021).The experiment was repeated three times.
Transwell assays
Cell migration and invasion were detected using the Transwell assay (Corning).For invasion assays, the upper chamber was precoated with 50 μL 20%Matrigel matrix.Cells were harvested and inoculated into the upper chamber in serum-free medium, and the lower chamber was filled with complete medium containing 20% fetal bovine serum as a chemotactic agent.Transwell plates were then incubated at 37 °C with 5% COfor 24 hours.The remaining cells were gently removed with sterile cotton swabs; the inserts were cleaned twice with PBS, fixed with 4% paraformaldehyde for 20 minutes, cleaned twice with PBS, stained with 0.3% hematoxylin for 20 minutes, and cleaned twice with PBS.The membrane was cut along the edge and sealed with neutral gum.Five randomly selected fields were photographed under a light microscope (Nikon) at 100× magnification and analyzed.
5-Ethynyl-2"-deoxyuridine assay
HRECs in logarithmic growth phase were seeded at 4 × 10to 1 × 10cells per well into a 96-well plate and cultured until they reached the normal growth phase.Cells were then treated as indicated in legends and incubated at 37°C and 5% COfor 24 hours.Next, reagent A from the Cell-LightEdu Apollo567 kit (KeyGen Biotech, Nanjing, China) was diluted with cell culture medium at 1:1000 for 5-ethynyl-2′-deoxyuridine (EdU) labeling, followed by incubation at 37°C, 5% COfor 2 hours.The cells were then fixed with 4% paraformaldehyde at room temperature for 30 minutes.Then, 100 μL of 2 mg/mL glycine solution was added to each well and decolorization was performed for 5–10 minutes on a horizontal shaker at room temperature; the glycine solution was discarded, and the cells were washed with PBS.Next,1× Apollo staining solution was prepared using the reagents following the kit instructions and 100 μL of 1× Apollo staining solution was added to each well;cells were then incubated at room temperature on a shaker in the dark for 30 minutes.The staining solution was discarded, cells were washed with PBS,and 4′,6-diamidino-2-phenylindole (Solarbio, Beijing, China) counterstaining was performed.The samples were observed and photographed under a fluorescence microscope.The number of stained nuclei of cells in the proliferation phase and the total number of nuclei were counted.Their ratio was calculated to compare the proliferation rate among groups.
Flow cytometry
Apoptosis was quantified by staining with the Annexin V-FITC/PI reagent(Vazyme Biotech, Nanjing, China).The cells were treated as indicated in legends.The samples were washed twice with PBS and centrifuged at 800×g
for 5 minutes to collect 1–5 × 10cells.The cells were suspended in 200 μL of binding buffer; 5 μL of Annexin V-fluorescein isothiocyanate (FITC) was added and mixed well, followed by the addition of 5 μL of propidium iodide (PI)counterstain.The samples were kept at 21°C in the dark for 10 minutes, and flow cytometry (BD, Franklin Lake, NJ, USA) was performed to determine cell apoptosis.Apoptosis quantification was performed in triplicate for all samples.Tube formation assay
On the day before tube formation assay, Matrigel matrix (Corning, Corning, NY,USA) was placed in a refrigerator at 4°C overnight.Once thawed, the Matrigel was centrifuged for 30 minutes and transferred onto ice for the subsequent steps.Matrigel was mixed with a pre-cooled pipette tip, and 500 μL aliquots were placed in pre-cooled Eppendorf tubes.Fifty microliters of Matrigel were added to each well of a pre-cooled 96-well plate, while preventing the formation of air bubbles.The samples were incubated at 37°C for 45 minutes.When the cells reached 70–80% confluence, they were harvested and resuspended in culture medium.Fifty microliters of resuspension medium were added to each well to obtain a concentration of 3 × 10/well; each condition was tested in triplicate.The samples were incubated at 37°C.After 4 hours, formed tubes were observed, photographed, and counted under a light microscope.
Animals and experimental groups
A total of 120 7-day-old C57BL6J mice (4.0–5.0 g) were used in experiments;mice were purchased from Changsheng Biology Co., Ltd.(Shenyang, China,license No.SCXK (Liao) 2015-0001).Animal housing and experimental procedures were performed at the Animal Laboratory in Shengjing Hospital,China Medical University.The housing environment was stable and specific pathogen free.The temperature was maintained at 23 ± 2°C, and the humidity was 52 ± 5%; animals were kept under a 12 hour light/dark cycle,with 6–8 animals in each cage.The experimental study was approved by the Animal Ethics Committee of Shengjing Hospital of China Medical University(approval No.2020PS078K, March 17, 2020).All experiments were designed and reported following the Animal Research: Reporting ofIn Vivo
Experiments(ARRIVE) guidelines (Percie du Sert et al., 2020).The animals were randomly divided into four groups: control, OIR, OIR +intravitreal injection of control lentivirus (OIR-C), and OIR + intravitreal injection of sh-piR-1245 lentivirus (OIR-T) groups (n
= 30 animals per group).In the control group, 7-day-old mouse pups and mothers were reared under standard conditions, without any treatment.For the other three groups, the pups and mothers were reared in a hermetically sealed glass container with an oxygen concentration of 75 ± 2% for 5 days; the container was opened once a day to change the bedding; fresh food and water were added, and the mother mouse was replaced (Figure 2).
Figure 2 | Schematic timeline of experimental groups in animal experiments.
For the OIR-C and OIR-T treatment groups, mice were removed from the oxygen box at 11 days old and anesthetized with 2% isoflurane (Woruide,Shenzhen, China) in a closed container.The pupils of both eyes were dilated,and the ocular surface was kept moist while intravitreal injection of PBS and piR-1245 knockdown lentivirus (1 μL each, 3E+8 TU/mL) was administered using a microsyringe.The needle was inserted 1 mm posterior to the limbus,taking care to avoid blood vessels.The bolus was slowly administered, and the needle was retained for 10 seconds before being swiftly removed.After application of gatifloxacin eye ointment (Xingqi, Shenyang, China) to the eyes to prevent endophthalmitis, mice were returned to the oxygen box.
After 5 days in the oxygen box (when mice were 12 days old), all mice were removed from the oxygen box and reared under normal conditions until they were 17 days old (Smith et al., 1994).Samples were collected for histopathological assays and total RNA and protein extraction.
To verify the effect of JAK2 inhibitor on RNV we further divided into OIR group, OIR-NC group and OIR-sh-p-JAK2 group (n
= 6 mice/group).In the OIR-sh-p-JAK2 group, 10 μM of the JAK2 inhibitor JAK2/TYK2-IN-1 (MCE,Shanghai, China) was injected into the vitreous cavity of 5-day-old mice with a microinjector (Hamilton, Reno, NV, USA).Retinal whole-mount preparation and blood vessel staining
To assess the extent of retinopathy, mice were anesthetized by 2% isoflurane in a closed container for 2 minutes; eyeballs were removed and fixed in 4%paraformaldehyde for 12 hours.The anterior segment of the eye and vitreous cavity contents were discarded, and the retina was carefully separated.The retina was incubated overnight with Isolectin B4–594 (Invitrogen) in a 4°C shaker away from light (Zhang et al., 2020a).Radial cuts were made in retinal tissue with the optic disc as the center, and the retina was spread out on a glass slide with the vitreous body facing upward.After adding antifade mounting medium, the slide was sealed with a cover slip, observed, and imaged under a fluorescence microscope.ImageJ software was used to calculate the area of non-perfusion area and neovascularization area.
Hematoxylin and eosin staining
To evaluate the pathological changes of RNV, the mice were anesthetized and sacrificed according to the methods described above, and eyeballs were removed.Samples were randomly selected from each group, fixed in 4%paraformaldehyde for 12 hours, and embedded; samples were cut along the sagittal plane of the optic nerve to form consecutive 4 μm-thick paraffin sections.Ten non-consecutive sections were obtained from each eye for hematoxylin and eosin (HE) staining, and sections containing the optic nerve were excluded.The HE staining work is to put the paraffin section into the automatic machine and seal them with neutral gum after dyeing.Images were captured using an optical microscope (Nikon); the numbers of nuclei from EC that had crossed the inner limiting membrane on the vitreous side (i.e.,neovascular nuclei) were counted, and the average number of neovascular nuclei was calculated for all slides.Only blood vessel nuclei that were closely adhered to the retina were counted; those not connected to the inner limiting membrane within the vitreous cavity were not included.
Real-time polymerase chain reaction
Total RNA was extracted from the cells and retinal tissues.The quality of total RNA was analyzed using an ultramicro ultraviolet spectrophotometer(Nano Drop, Thermo, Waltham, MA, USA).High-quality RNA samples were selected for reverse transcription using the PrimeScriptRT Master Mix reverse transcription kit (Takara, Shiga, Japan) following the manufacturer’s instructions.The 10 μL reaction mixture included 500 ng of template RNA and 2 μL of Master Mix, and the remaining volume was DNase/RNase-Free Water (Solarbio) After evenly mixing the three components, the sample was placed in a 37°C water bath for 15 minutes and then transferred to an 85°C water bath for 5 minutes to terminate the reverse transcriptase activity,yielding the cDNA amplification template.Real-time polymerase chain reaction (qPCR) was performed using SYBR®Premix Ex Taqfluorescent dye (Takara).Each well contained 20 μL of reaction mixture, including 10 μL of SYBR, 0.4 μL of 50× Passive Reference Dye, 2 μL of cDNA, and 1 μL of primers (forward and reverse, 0.5 μL each); the remaining volume was ddHO.The reaction mixture was loaded onto a 7500 Plus Real Time PCR system (ABI, Carlsbad, CA, USA).The PCR protocol was as follows: 15 seconds at 95°C for 40 cycles and 30 seconds at 60°C.U6 was used as the housekeeping gene to normalize piR-1245 expression data, and β-actin mRNA was used to normalize VEGF expression.Data were analyzed using the 2method (Wang et al., 2022).All procedures were performed on ice using RNAse-free Eppendorf tubes and pipette tips.Primers are listed in Table 1.

Table 1 |Sequence of PCR primers
Western blot assay
Total protein was extracted from cells and retinal tissues by radioimmunoprecipitation assay (Solarbio), and the protein concentration of samples was determined using the bicinchoninic acid assay kit (Beyotime, Shanghai,China).Protein loading buffer was added at a ratio of 4:1; the sample was mixed evenly, denatured in boiling water for 5 minutes, and centrifuged to obtain the supernatant.Protein samples (30 μg) were separated by sodium dodecyl sulfatepolyacrylamide 10% gel electrophoresis and transferred onto a polyvinylidene difluoride membrane.Membranes were blocked with 5% non-fat dry milk for 1 hour and washed with Tris-buffered saline with 0.1% Tween® 20 Detergent.Membranes were then incubated with primary antibodies (all 1:1000-diluted,polyclonal, rabbit: Bax antibody, Cat# 50599-2-Ig, RRID: AB_2061561; Bcl-2 antibody, Cat# 16026-1-AP, RRID: AB_2827646; caspase-3 antibody, Cat#19677-1-AP, RRID: AB_10733244; STAT3 antibody, Cat# 10253-2-AP, RRID:AB_2302876; VEGF antibody, Cat# 19003-1-AP, RRID: AB_2212657; hypoxia inducible factor-1α (HIF-1α) antibody, Cat# 20960-1-AP, RRID: AB_10732601;interleukin-1 (IL-1β) antibody, Cat# 66737-1-Ig, RRID: AB_2882087; IL-6 antibody, Cat# 21865-1-AP, RRID: AB_11142677; and tumor necrosis factor-α(TNF-α) antibody, Cat# 17590-1-AP, RRID: AB_2271853; Proteintech, Chicago,IL, USA; cleaved Caspase-3, Cat# ab2303, RRID: AB_302962; JAK2 antibody,Cat# ab108596, RRID: AB_10865183; phosphorylated STAT3 antibody, Cat#ab76315, RRID: AB_1658549; phosphorylated JAK2 antibody, Cat# ab32101,RRID:AB_775808, Abcam, Cambridge, UK) at 4°C overnight.The membrane was washed three times (5 minutes each) with Tris-buffered saline with 0.1% Tween and incubated at 37°C for 2 hours in secondary antibody solution (horseradish peroxidase-labeled goat anti-rabbit IgG, 1:500, Proteintech, Cat# SA00001-2; RRID: AB_2722564).The membrane was developed in a dark room using an enhanced chemiluminescence kit (Solarbio), and images were acquired using a gel imaging system (ABI).Band grey values were analyzed using the ImageJ analysis software.β-Tubulin (1:1000, polyclonal, rabbit, Cat# YM3030,ImmunoWay, Plano, TX, USA) served as the internal control for expression normalization.
Statistical analysis
No statistical methods were used to predetermine sample sizes; however, our sample sizes are similar to those reported in a previous publication (Wang et al., 2022).The number of experimental animals in each group was six biological replicates.All experiments were performed in triplicate.No animals or data points were excluded from the analysis.Statistical analysis was conducted using SPSS 22.0 (IBM, Armonk, NY, USA).All statistical data are expressed as the mean ± standard deviation (SD).Between-group differences were tested using one-way analysis of variance analysis of variance followed by Bonferroni’spost hoc
test.P
< 0.05 was considered statistically significant.The experiments were repeated three times.Random double-blind counting was used.Results
piR-1245 has no effect on proliferation and apoptosis of HRECs in normoxic conditions
To study the effect of piR-1245 on the function of HRECs cells under normoxia,we knocked down piR-1245 of HREC cells and observed it under normoxia.The expression level of piR-1245 in the sh-piR-1245 group was lower than that in the sh-NC group (P
< 0.01), and the protein expression level of PIWIL2 in the sh-piR-1245 group was lower than that in the sh-NC group (P
< 0.05;Figure 3A–C).The results of flow cytometry showed that under normoxia, there was no significant change in the apoptosisof HREC in response to piR-1245 knockdown.The results of Transwell assay and scratch assay showed that under normoxia, there was no significant change in the migration of HREC in response to piR-1245 knockdown.The results of EdU assay showed that under normoxia, there was no significant change in the proliferation of HREC in response to piR-1245 knockdown (allP
> 0.05; Figure 3A–K).piR-1245 knockdown inhibits the proliferation, migration, and tube formation of HRECs under hypoxic conditions
RNV is mainly due to ischemia and hypoxia, so we studied the pathophysiological changes of HRECs under hypoxia.The expression levels of piR-1245 and PIWIL2 in HRECs of hypoxia and hypoxia + sh-NC groups were significantly higher than those in the hypoxia + sh-piR-1245 and control groups (allP
< 0.05; Figure 4A–C).Flow cytometry results indicated that the apoptosis rate of the hypoxia group was significantly higher than that of the control group (P
< 0.05).Furthermore, the apoptosis rate was significantly lower in the hypoxia + shpiR-1245 cells than that in hypoxia + sh-NC cells (P
< 0.05; Figure 4D and E).A migration scratch assay revealed that the migration rate of the hypoxia group was significantly higher than that of the control group (P
< 0.05).Additionally,the migration rate of the hypoxia + sh-piR-1245 group was significantly lowerthan that of the hypoxia + sh-NC group (P
< 0.05; Figure 4F and G).The results of the Transwell assay were in line with those of the scratch assay (Figure 4H and I).Tube formation assay showed that number of tubes formed by the hypoxia group was significantly higher than that in the control group (P
< 0.05),while significantly fewer tubes were formed in the hypoxia + sh-piR-1245 group than in the hypoxia + sh-NC counterparts (P
< 0.05; Figure 4J and K).EdU assay showed that the number of proliferating cells in the hypoxia group was significantly higher than that in the control group (P
< 0.05), while fewer proliferating cells were observed in the hypoxia + sh-piR-1245 group than in the hypoxia + sh-NC group (P
< 0.05; Figure 4L and M).piR-1245 knockdown inhibited the proliferation, migration, and tube formation of HRECs under hypoxic conditions.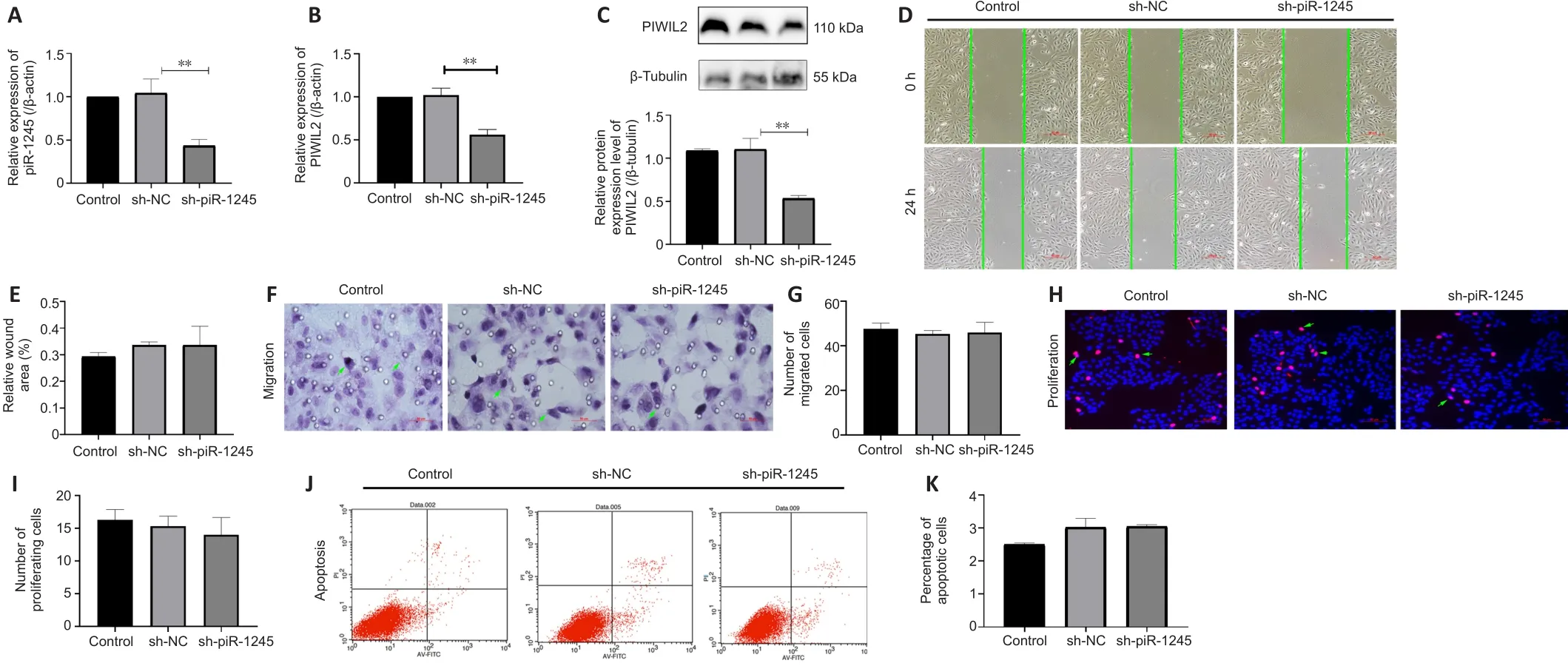
Figure 3 | piR-1245 has no effect on HRECs in normoxic conditions.
piR-1245 regulates HIF-1α and VEGF expression in HRECs through the JAK2/STAT3 pathway in the hypoxic condition
The expression levels of p-JAK2, p-STAT3, HIF-1α and VEGF were increased in HRECs in hypoxic culture conditions.To investigate the effect of pir-1245 on AK2/STAT3 pathway and whether JAK2/STAT3 had a targeted regulatory effect on VEGF, we knocked down piR-1245 in hypoxic-treated cells and examined the expression levels of p-JAK2, p-STAT3, HIF-1α and VEGF by western blotting.The expression levels of p-JAK2, p-STAT3, HIF-1α and VEGF were significantly reduced upon piR-1245 knockdown compared with levels in the hypoxia control groups (allP
< 0.05; Figure 5A–D).To verify the effects of piR-1245 on JAK2/STAT3 pathway and the targeted regulation of JAK2/STAT3 on VEGF, cells were interfered with p-JAK2 activator.qPCR results showed that while the level of piR-1245 was reduced in hypoxic cells downregulated for piR-1245 (P
< 0.05), piR-1245 levels did not change with additional treatment of the p-JAK2 activator compared with levels in the control (sh-NC-p-JAK2 activator) group (P
> 0.05; Figure 6A).Western blot assay showed that while the expression levels of HIF-1α and VEGF in the hypoxia-sh-piR-1245 group were significantly lower than those in the hypoxia group (bothP
< 0.05), there was no difference between hypoxiash-piR-1245-p-JAK2 activator and hypoxia-sh-NC-p-JAK2 activator groups (bothP
> 0.05; Figure 6B and C).piR-1245 knockdown inhibits RNV in vivo
We next examined the role of piR-1245 on RNV using a mou se model of oxygen-induced retinopathy (OIR; Figure 2).Mice were divided into four treatment groups: control, OIR, OIR + intravitreal injection of control lentivirus(OIR-C), and OIR + intravitreal injection of piR-1245 knockdown lentivirus(OIR-T).The degree of retinopathy in mouse tissue was assessed using retinal wholemount staining and HE staining (Figure 7A–F).Retinal wholemount staining indicated that the retinal tissues of control and OIR-T mice exhibited a clear and regular blood vessel course, without bleeding points or leaks.OIR and OIR-C retinal tissues had tortuous and occluded retinal blood vessels,bleeding and leakage, NV clusters, and large non-perfused areas.The nonperfused area and NV area in the OIR group was significantly greater than those in the control group (P
< 0.05).Furthermore, the non-perfused area and NV area of OIR-C samples were significantly greater than in OIR-T group (bothP
< 0.05).HE staining revealed that the number of EC nuclei that had crossed the inner limiting membrane on the vitreous side was significantly greater in the OIR group than that in the control group (P
< 0.05).Furthermore, the area of neovascular nuclei in the OIR-C group was significantly greater than that in the OIR-T group (P
< 0.05).Western blot assay was performed to determine the protein levels of inflammatory factors in retinal tissue.While expression levels of IL-1β, IL-6, and TNF-α were significantly higher in the OIR group than those in the control group (allP
< 0.05), these levels were significantly lower in the OIR-T group than those in the OIR-C group (allP
< 0.05; Figure 7G–I).Thus, the OIR-T group exhibited a significantly suppressed inflammatory response.Additionally, while cleaved caspase-3 and Bax levels were significantly higher and Bcl-2 was significantly lower in the OIR group than those in the control group (P
< 0.05), cleaved caspase-3 and Bax levels were significantly lower and Bcl-2 was significantly higher in the OIR-T group than those in the OIR-C group (P
< 0.05).Thus, the OIR-T group exhibited a significantly suppressed inflammatory response.These data indicated that intravitreal injection of piR-1245 knockdown lentivirus suppressed the inflammatory response and reduced retinal tissue apoptosis in the OIR model.We further found that while expression levels of p-JAK2, p-STAT3, HIF-1α,and VEGF were significantly higher in the OIR group than those in the control group (allP
< 0.05), these levels were significantly lower in the OIR-T group than those in the OIR-C group (allP
< 0.05) (Figure 7J–K).Thus, the OIR-T group exhibited a significantly suppressed inflammatory response in retinal blood vessels through the JAK2/STAT3 pathway.p-JAK2 inhibitor inhibits RNV in vivo
The mice in the OIR + sh-p-JAK2 group were intravitreally injected with p-JAK2 inhibitor, and the mice in the OIR-NC group were used as a control.The expression levels of p-JAK2, p-STAT3, HIF-1α, and VEGF were significantly lower in the OIR + sh-p-JAK2 group than those in the OIR and OIR-NC groups(allP
< 0.05; Figure 8).This result indicates that p-JAK2 inhibitor can regulate VEGF and HIF-1in vivo
α which in turn inhibits the generation of RNV.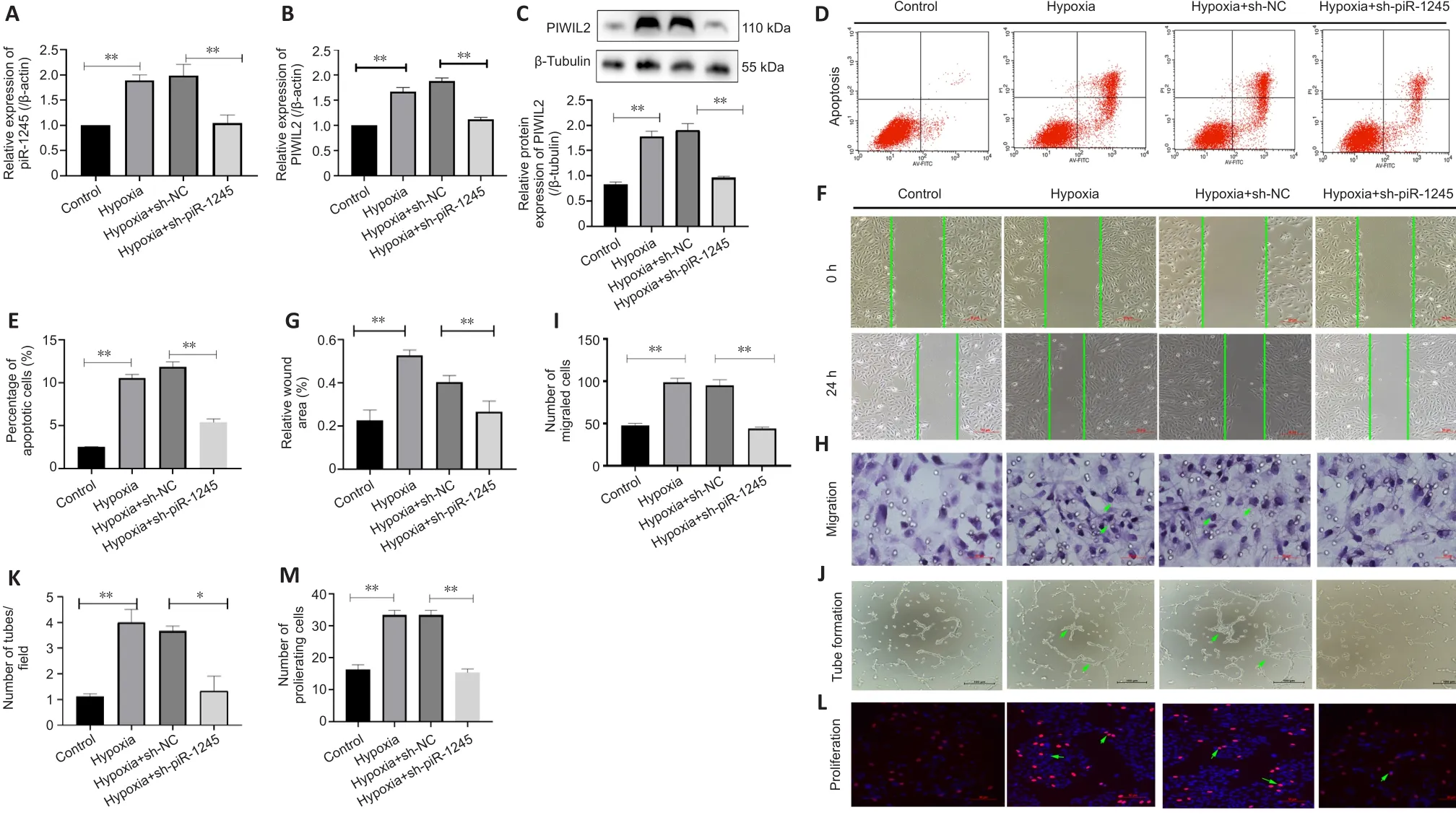
Figure 4 | piR-1245 knockdown inhibits HREC apoptosis, proliferation, migration and tube formation in hypoxic conditions.

Figure 5|piR-1245 regulates the expression of HIF-1α and VEGF in HRECs through JAK2/STAT3 pathway in the hypoxic condition.
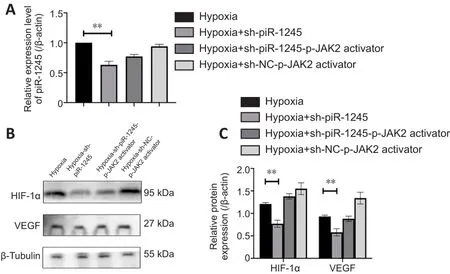
Figure 6|piR-1245 regulates HIF-1α and VEGF expression in HRECs through the JAK2/STAT3 pathway in the hypoxic condition.

Figure 7| Intravitreal injection of piR-1245 inhibits RNV, regulates JAK2/STAT3 activation and VEGF expression in the retina.
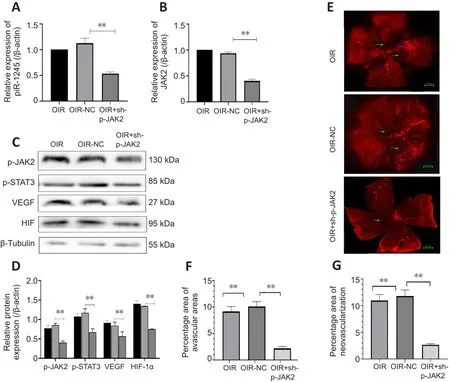
Figure 8| Intravitreal injection of p-JAK2 inhibitor relieves RNV.
Discussion
RNV is the main underlying cause of blindness across all age groups.Common RNV-related ocular diseases include retinopathy of prematurity, diabetic retinopathy, and central retinal vein occlusion (Peng et al., 2020; Vähätupa et al., 2020).NV involves the proliferation, migration, and sprouting of ECs and is regulated by a variety of complex signaling pathways involving cytokines,adhesion molecules, proteases, and other molecular factors (Bhowmik et al., 2021; Sato et al., 2021).RNV-related ocular diseases are associated with large areas of hypoxia and ischemia in the retina, leading to microcirculatory disorders and disrupted homeostasis (Sui et al., 2020).This imbalance results in the aberrant production of angiogenic growth factors, such as VEGF, and incomplete development of new blood vessels may lead to leakage and hemorrhage throughout vessel growth (Arya et al., 2021; Masłowska et al.,2021).This causes serious complications, such as retinal detachment or vitreous hemorrhage, eventually leading to the destruction of photoreceptor cells and loss of vision.
Angiogenesis is a complex pathological process regulated by a multitude of factors and its molecular and cellular mechanisms remain poorly understood(Novy et al., 2021; Zhang et al., 2021b).The interactions of ECs, pericytes, and astrocytes within retinal blood vessels jointly regulate retinal angiogenesis(Smolders et al., 2021).The main pathological change observed in RNV is the formation of new blood vessels, driven by a wide range of angiogenic factors,including VEGF (Bredow et al., 2007).In this study, the expression levels of inflammatory factors IL-1, IL-6 and TNF-α were remarkably high in the retina of the OIR mouse model.After we knocked down piR-1245, the expression of inflammatory factors decreased.The preceding statements suggest that piR-1245 is involved in regulating the expression of inflammatory factorsbut does not address an involvement of the factors in RNV.
The majority of transcripts derived from the human genome are non-coding RNAs, which can be classified into short non-coding RNAs and long non-coding RNAs (Boudewijn et al., 2020).Short non-coding RNAs include microRNAs,siRNAs, and piRNAs (Kumar et al., 2020; Tamtaji et al., 2020).The biological action of piRNAs is different from those of siRNAs and miRNAs (Siddiqi and Matushansky, 2012; Beltran et al., 2021; Xie et al., 2022).The latter two mainly exert their gene silencing effects by interacting with the Ago protein subfamily, while piRNAs bind with PIWI and form a piRNA complex to silence expression (Zhou et al., 2021).As a newly discovered gene regulatory factor,piRNA has been found to regulate gene expression at the pre-transcriptional,post-transcriptional, and epigenetic levels, hence playing a vital role in the development of organisms (Clark et al., 2017; Zhang et al., 2018).Studies have shown that piRNAs are involved in regulating the maturation of male germ cells and the maintenance of genomic stability, and PIWI protein prevents the transposition of testicular germline retrotransposons (Amaar and Reeves,2020; Xin et al., 2021).Furthermore, in the early stages of Drosophila embryo differentiation, PIWI proteins positively regulate the translation process,allowing piRNAs to promote the stability and translation of their target mRNA molecules (Ramat and Simonelig, 2021).piRNAs inhibit retrotransposons,maintain DNA integrity, directly promote repair, the aggregation of chromosomal structures against damage, and inhibit DNA damage-causing autosomal gene expression.In this study, we found that PIWIL2 and piR-1245 were highly expressed in the retinal tissues of OIR mice and hypoxic HRECs.Additionally, piR-1245 knockdown in hypoxic HRECs inhibited VEGF expression and reduced apoptosis, migration, and tube formation.Intravitreal injection of piR-1245 knockdown lentivirus into OIR mice suppressed the inflammatory response and inhibited the development of pathological NV.These results may provide new directions for the treatment of RNV-related retinopathies.
The JAK2/STAT3 cascade is a major signaling pathway that regulates a variety of cellular functions, such as cell proliferation, differentiation, and transformation(Yu et al., 2019; Di et al., 2020; Zhang et al., 2020b).It can also promote NV and the recruitment of inflammatory cells.STAT3 promotes cell proliferation,and its sustained activation causes uncontrolled proliferation and enhanced cell migration.To explore the role of piR-1245 in HRECs and its relationship with JAK/STAT3 signaling, we performed piR-1245 knockdown and JAK inhibitor treatment.piR-1245 knockdown and JAK inhibition both suppressed JAK/STAT3 signaling, reducing VEGF expression.We found that pir-1245 could further activate JAK2/STAT3 by regulating the phosphorylation levels of JAK2 and STAT3 proteins in JAK2/STAT3.The observed levels of p-JAK2, p-STAT3, HIF-1, and VEGF support the conclusion about the relation between RNV and JAK2/STAT3 signaling.This study is the first to propose that PIWIL2/piR-1245 regulates the biological behavior of retinal ECs and RNV development via the JAK/STAT3/VEGF pathway.Our findings can serve as a reference for the selection of therapeutic targets in RNV-related diseases.However, this study has certain limitations.We examined this pathway in cellsin vitro
, and the sample size of animalsin vivo
is insufficient.Further studies usingin vivo
models and with a larger sample size are required.Future clinical research will be useful to investigate potential new therapeutic treatment methods.Author contributions:
YY and XLC designed this study, conducted the experimental analysis, and wrote the manuscript.XLC, YD, QZN participated in study design, statistical analysis, data analysis and manuscript preparation.XLC revised the manuscript.All authors have read and approved the final manuscript.
Conflicts of interest:
The authors declare that they have no conflict of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Vanessa Castelli, University of L’Aquila, Italy.
Additional file:
Open peer review report 1.
- 中国神经再生研究(英文版)的其它文章
- Patient-specific monocyte-derived microglia as a screening tool for neurodegenerative diseases
- Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases
- Inflammation in diabetic retinopathy: possible roles in pathogenesis and potential implications for therapy
- Targeting the nitric oxide/cGMP signaling pathway to treat chronic pain
- Neurosteroids as stress modulators and neurotherapeutics: lessons from the retina
- Myelinosome organelles in pathological retinas:ubiquitous presence and dual role in ocular proteostasis maintenance