
共价有机骨架/碳纳米管复合材料中锂离子吸附与传输特性的分子模拟
2022-12-02 11:55徐毅崔致远吴凡袁彬
上海大学学报(自然科学版) 2022年1期
徐毅,崔致远,吴凡,袁彬
(1.上海大学环境与化学工程学院,上海 200444;2.魏德曼检测技术(上海)有限公司,上海 201114)
随着现代社会的快速发展,人类对能源的需求与日俱增,而煤炭、石油、天然气等不可再生能源日益减少,人们将更多的目光聚焦于新能源的开发,因此与之相关的储能系统受到广泛关注.锂电池作为电化学储能系统的代表显得尤为突出,并成为了2019诺贝尔化学奖的“主角”.
锂电池内部的电极材料至关重要,因而相应的遴选标准也较为严苛.目前,单一物质往往难以满足相关标准,故可能产生协同效应的二元或多元复合材料自然应运而生.近年来,以金属有机骨架(metal organic framework,MOF)和共价有机骨架(covalent organic framework,COF)为代表的有机骨架化合物与碳纳米管(carbon nanotube,CNT)所形成的复合材料COF@CNT逐渐进入人们的视线.现有的研究结果充分表明,复合材料COF@CNT可将MOF、COF的多孔、高比表面积等优点与CNT的强导电性、高模量、高强度等优势有机融合,从而拥有诸多优良的性能,应用领域遍及新能源材料、电催化、电传感、电磁、气体捕集和环境保护等,而其在各类电极材料中的应用则尤为突出.2013年,Wang等[1]合成了均苯三甲酸铜Cu3(BTC)2与多壁碳纳米管(multi-walled carbon nanotube,MWCNT)的复合材料,并利用其修饰电极.通过基于纳米氧化锌前驱体的纳米管促进生长,Yue等[2]制备了咪唑分子筛骨架(zeolite imidazolate framework,ZIF)与MWCNT的复合材料,该复合材料在0.1°C的锂-硫电池中体现出优良的性能.2015年,Xu等[3]将一种具备氧化还原活性的晶体状介孔COF与CNT进行复合后用作电极材料,制备的锂离子电池具有高效率、强循环稳定性和高倍率性能等优点.Zhang等[4]将合成的锰基MOF与CNT的复合材料用于制备超级电容器的电极,获得了导电性能的内在提升和比电容的内在增加.Wen等[5]发现镍基MOF的特殊结构与CNT的高导电率所产生的协同效应使复合材料MOF@CNT具有卓越的电化学性能.2017年,Yang等[6]制备了一种铁基MOF、CNT和石墨烯共同构成的复合材料,并发现由其作为电极组装的锌-空气电池具有较低的充放电过电位.Han等[7]将TpPa-COF(Tp为1,3,5-三甲酰基间苯三酚,Pa为对苯二胺)以纳米尺度包覆于单壁碳纳米管(single-walled carbon nanotube,SWCNT)制得复合材料,应用该复合材料制备的超级电容器具有较高的比电容与循环稳定性.借助于4,4′,4′′-(1,3,5-三嗪-2,4,6-三基)三苯胺(4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline,TTA)和2,5-二羟对苯二酚醛(2,5-dihydroxyterepthaldehyde,DHTA)的共价连接,Sun等[8]制得了具有高度有序孔隙通道的COF与氨基官能化的MWCNT共同组成的复合材料,该复合材料具有更为优异的电化学性能.Mao等[9]开发了一种基于MOF和CNT的多级孔结构柔性复合薄膜,用其制备的高性能可折叠柔性锂-硫电池具有优异的循环性能、较高的硫载量和能量密度,并可在不同折叠程度上保持稳定的能量输出,从而为高能量密度和柔性的锂-硫以及锂离子、钠离子和锂-空气电池等提供了新的设计思路[10].2018年,Lei等[11]将二维COF卷曲包裹于CNT表面制备了复合材料,用其作为负极的的锂电池获得了较高的可逆容量,他们还利用第一性原理计算揭示了该复合材料的储锂机理.Xu等[12]将合成的MOF-74-Ni/CNT复合材料用作锂-硫电池中的硫载体(阴极),制备的锂-硫电池具有较高的初始放电容量、良好的倍率性能和较低的容量衰减.通过在导电MWCNT上生长多孔TpPa-COF,Zhang等[13]制备了一种新型的具有核壳结构的纳米复合材料,实验结果表明,用其制备的电池具有较高的初始放电容量和优异的容量保持能力.Pu等[14]合成了一种新型的UiO-66/CNT复合材料,该复合材料具有优越的结构稳定性、高导电性和对于多硫化物的强化学吸附能力,因而在锂-硫电池中可作为具有稳定循环性能的硫阴极.第一性原理计算和表观吸收的结果表明,UiO-66/CNT对可溶性多硫化物具有明显的包封作用,从而使得S@UiO-66/CNT电极拥有出色的循环性能.最近,Chen等[15]将硼酯基COF材料成功包覆于CNT外表面,并将其运用于钾离子电池的阴极,从而使其拥有更多暴露的活性位点、更快的钾离子传输动力学以及更高的钾离子存储性能.章琴等[16]制备了TpPa-COF材料,并将其与导电性能优异的MWCNT进行复合,所得材料制备的锂电池表现出良好的大电流充放电性能.为提升多金属氧酸盐基MOF材料的导电性能,Li等[17]在其晶体结构中引入了功能化的SWCNT,并将所得复合材料应用于锂电池的负极,制备的锂电池获得了较高的可逆容量.
综上所述可知,通过对有机骨架化合物与CNT各自拥有的优势“做加法”,可产生相应的协同效应,由它们共同组成的复合材料能够展现出更为优良的电化学性能.因此,对此开展基础性研究有助于从微观层面上对该复合材料电化学行为的内在机理进行深入理解与认识,并在构筑具有复合结构的新能源材料方面发挥重要的作用.但就目前的情况来看,与之相关的研究工作既不全面,也不深入,且基本局限于实验研究,而真正能够对其内在机理进行深入探寻并可将结果直观呈现的理论研究鲜有报道[11,14,18].为此,本工作以分子模拟为主要研究手段,以COF@CNT为研究对象,探索Li+在该材料中的吸附与传输特性,为进一步提升此类物质的应用价值、挖掘其应用潜力和扩展其应用领域提供必要的理论指导.本工作中,分子模拟均利用Materials Studio(MS)软件中的Forcite模块来完成.
1 模型构造
1.1 单体模型
将具有二维层状结构且包含亚胺键的COF-LZU1[19]作为共价有机骨架的研究对象,并将其单位晶胞作为COF单体,而CNT单体则选取7个单位长度(1.722 nm)的(6,6)型单壁碳纳米管.单体的分子模型如图1所示.
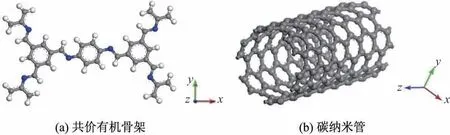
图1 COF和CNT单体的分子模型Fig.1 The molecular model of monomers COF and CNT
1.2 复合体模型
为获得相关实验研究中的包覆结构[11],首先将两个COF单体分别置于一个CNT单体的两侧,并使前者的长轴与后者的主轴相互垂直,即可得到COF@CNT的初始模型;然后,以该模型为基础,建立相应的晶胞,其晶格参数为:a=b=6 nm、c=2 nm、α=β=90°、γ=120°.体系中的所有原子都被允许在3个方向上完全弛豫,优化的能量收敛公差为2×10-5kcal/mol,力收敛标准为0.01 kcal/(mol·nm-1),位移收敛标准为1.0×10-4nm,力场与电荷分别选取为Universal与Charge using QEq,对其实施几何优化,最终得到如图2所示的分子结构(晶格未显示).显然,COF已完全包覆于CNT之上,此时整体能量最低,故将该结构作为COF@CNT的分子模型用于后续研究.
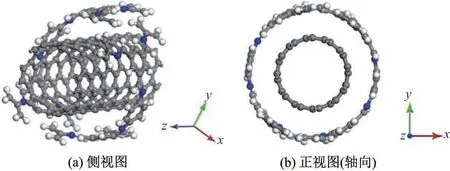
图2 COF@CNT分子模型(优化后)Fig.2 Molecular model of COF@CNT(optimized)
2 模拟结果与讨论
根据相关的实验研究结果[11]可知,对于Li+,CNT能够提供的吸附位点和吸附量远不如COF,故本工作将Li+的吸附位点均集中于后者,以便在最大程度上探寻COF@CNT对于Li+的吸附特性.
2.1 Li+的吸附位点与吸附顺序
Li+在COF单体上的吸附位点如图3所示.基于COF单体本身的结构特征,Li+的吸附位点可分为5类:氮位(1);双支链苯环的非支链位(2);三支链苯环的支链位(3);三支链苯环的非支链位(4);双支链苯环的支链位(5).
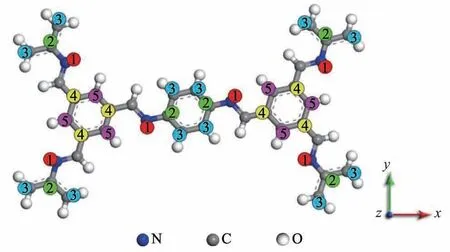
图3 Li+在COF单体上的吸附位点Fig.3 Adsorption sites of Li+on the COF monomer
将Li+分别吸附于上述5类位点,并对COF@CNT再次进行几何优化,所得吸附键长与键能如表1所示.可以发现:若按吸附键长递增的顺序,吸附位点排列依次为1,2,3,4,5;而与之相对应的吸附键能则恰好呈递减关系.这主要是因为吸附键越长,其键能越小.

表1 各类吸附位点的吸附键长与键能Table 1 Length and energy of the adsorption bond on each type of site
同等条件下,键能越大的位点对于金属离子应更具吸引力.因此,Li+在COF@CNT上的吸附过程可分为如下5个阶段:①12个Li+吸附于位点1,Li+的吸附总数为12;②12个Li+吸附于位点2,Li+的吸附总数为24;③24个Li+吸附于位点3,Li+的吸附总数为48;④12个Li+吸附于位点4,Li+的吸附总数为60;⑤12个Li+吸附于位点5,Li+的吸附总数为72.
2.2 Li+的吸附能
对于吸附行为而言,吸附的难易程度可用吸附能来进行衡量.Li+与COF@CNT的吸附能为

式中:Ea表示Li+的平均吸附能;E0表示未结合Li+的COF@CNT体系能量;ELi+为单个Li+的能量;ET则为吸附Li+后的COF@CNT体系总能量;n是Li+的吸附数量.显然,Ea为正,表示COF@CNT吸附Li+这一行为在热力学上是有利的;Ea越大,所得结构越稳定.
Li+在COF@CNT中的吸附数量n对吸附能Ea的影响如图4所示.由图4可以看出:当n=12时,吸附处于第1阶段,此时氮位对Li+的吸附键能最高,故Li+发生脱附的可能性最小,体系最为稳定,Ea最大;随着n不断增大,吸附进入第2~5阶段,后续位点对Li+的吸附键能逐渐减小,故Li+发生脱附的可能性逐渐增大,从而导致Ea逐渐下降;2.01 eV时,吸附能Ea仍然大于Li2的内聚能(0.94 eV)和固体Li的内聚能(1.63 eV)[20].由此可以推测,相较于相互聚集,Li+更倾向于被COF@CNT所吸附,而这无疑有利于提高该复合材料的锂化效率,增加其锂容量,从而使其有望成为锂电池的电极材料.

图4 Li+在COF@CNT中的吸附数量对其吸附能的影响Fig.4 Dependence of the adsorption energy of Li+on its adsorption number in COF@CNT
2.3 COF@CNT的形貌变化
复合材料的形貌对于金属离子在其中的吸附与传输具有较大影响,因而值得重点关注.Li+的吸附过程中,COF@CNT的形貌变化如图5所示.显然,在初始状态下,两个COF单体以“头尾相连”的方式组成闭环结构包覆于CNT之上;而随着Li+吸附数量的不断增加,该闭环结构逐渐被打破.作为吸附行为的主、客体,COF与Li+均逐渐远离CNT(见图6),其主要原因在于,随着Li+的不断吸附,COF与CNT之间占主导地位的π-π堆积作用逐渐减弱,从而使得二者之间的作用力减小,距离自然增大.此外,从氮位饱和吸附(见图5(b))至整体饱和吸附(见图5(f))的过程中,N-Li键的长度始终保持在0.195 nm,充分表明氮位点的吸附不受其他位点吸附状态的影响,同时也再次说明该位点的吸附稳定性最强.
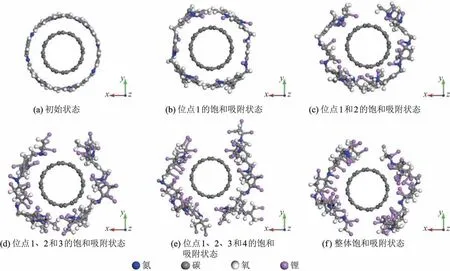
图5 COF@CNT在Li+吸附过程中的形貌变化Fig.5 Morphology change of COF@CNT during the adsorption process of Li+
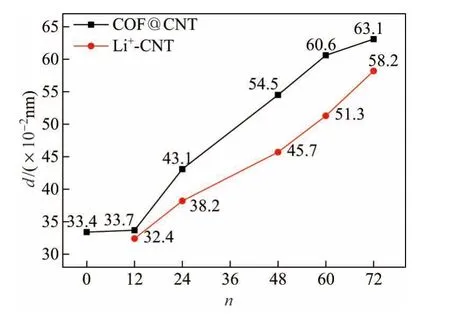
图6 COF@CNT、Li+-CNT间距与Li+吸附数量的关系Fig.6 Dependence of COF@CNT distance and Li+-CNT distance on the adsorption number of Li+
2.4 COF@CNT的体积变化
为了保证锂离子电池的安全性和稳定性,其电极材料需要在充/放电循环期间维持较低的体积变化率.COF@CNT的体积变化率为

式中:VmT和Vm0分别表示已结合与未结合Li+的COF@CNT体积.
图7为COF@CNT的体积变化率η与Li+吸附数量n的关系.显然,随着n增加,η不断增大.在吸附Li+的过程中,COF逐渐远离被其包覆的CNT,从而导致COF@CNT的体积不断增大.当达到饱和吸附状态时(n=72),η最大,但仅为0.25.可见,COF@CNT在锂化过程中体积变化较小,能够满足锂电池中电极材料的基本要求.
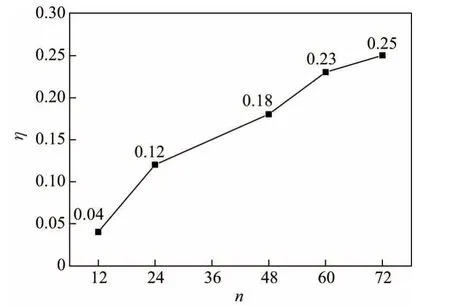
图7 COF@CNT的体积变化率与Li+吸附数量的关系Fig.7 Relationship between the volumetric change rate of COF@CNT and the adsorption number of Li+
2.5 平均电压与理论容量
电压是锂离子电池的一个重要参数.理想的电极材料应同时具有电压足够低的负极平台和电压足够高的正极平台.若将COF@CNT作为电极材料,其平均电压为[21–22]

式中:ΔG为Gibbs自由能;F为Faraday常数;n是Li+的吸附数量.

式中:PΔV是体积效应;TΔS是熵效应[23];ΔE为因Li+吸附而导致的体系内能变化,

相对于ΔE,PΔV和TΔS均可忽略不计,则有
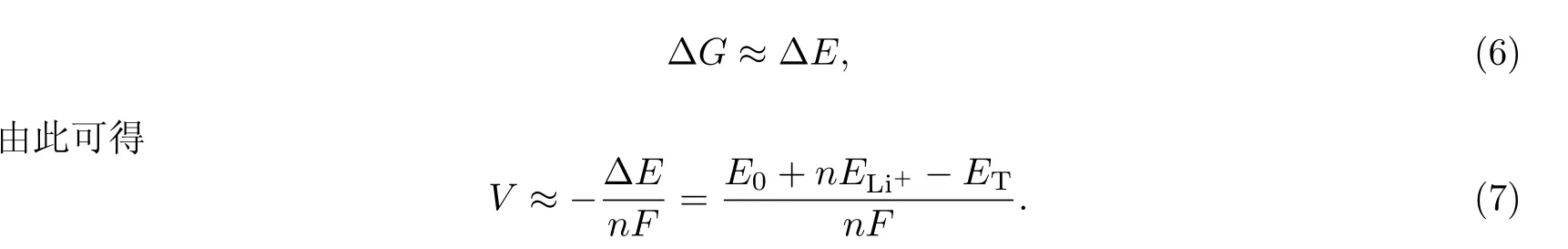
式(7)已被广泛用来计算平均电压[20,24].若能量以eV为单位,则最终的计算结果与式(1)所得Ea数值完全相等.
COF@CNT的平均电压V与Li+吸附数量n的关系如表2所示.显然,随着n增加,V逐渐降低.当n达到最大时,V仍可保持在2.00 V以上,再次说明COF@CNT适合作为锂电池的电极材料.
众所周知,锂电池的储锂容量对其实际应用至关重要.COF@CNT作为电极材料时,估算得到的理论容量为:

式中:ne为充放电过程中涉及的电子数;F是法拉第常数;M为COF@CNT的单位晶胞质量.
表2为COF@CNT的平均电压和理论容量.可以发现,Ct随着n的增加而增加,最大可达1 402.47 mAh·g-1,再次表明COF@CNT有望成为锂电池的电极材料.

表2 COF@CNT的平均电压、理论容量与Li+吸附数量的关系Table 2 Dependence of the average voltage and the theoretical capacity of COF@CNT on the adsorption number of Li+
2.6 Li+的电导率
为探索Li+在COF@CNT中的传输特性,本工作对其进行分子动力学模拟,主要参数如下:晶格参数a=b=4 nm,c=2 nm,α=β=γ=90°;正则系综(canonical ensemble,NVT);体系温度T=298 K(Velocity Scale热浴);时间步长Δt=1 ps,总时长t=4 ns.
根据所处位置的不同,Li+在COF@CNT中的传输路径(见图8)可分为3种:CNT内部,CNT与COF之间,COF外部,与之相对应的Li+在传输过程中的均方位移(mean square displacement,MSD)统计结果如图9所示.
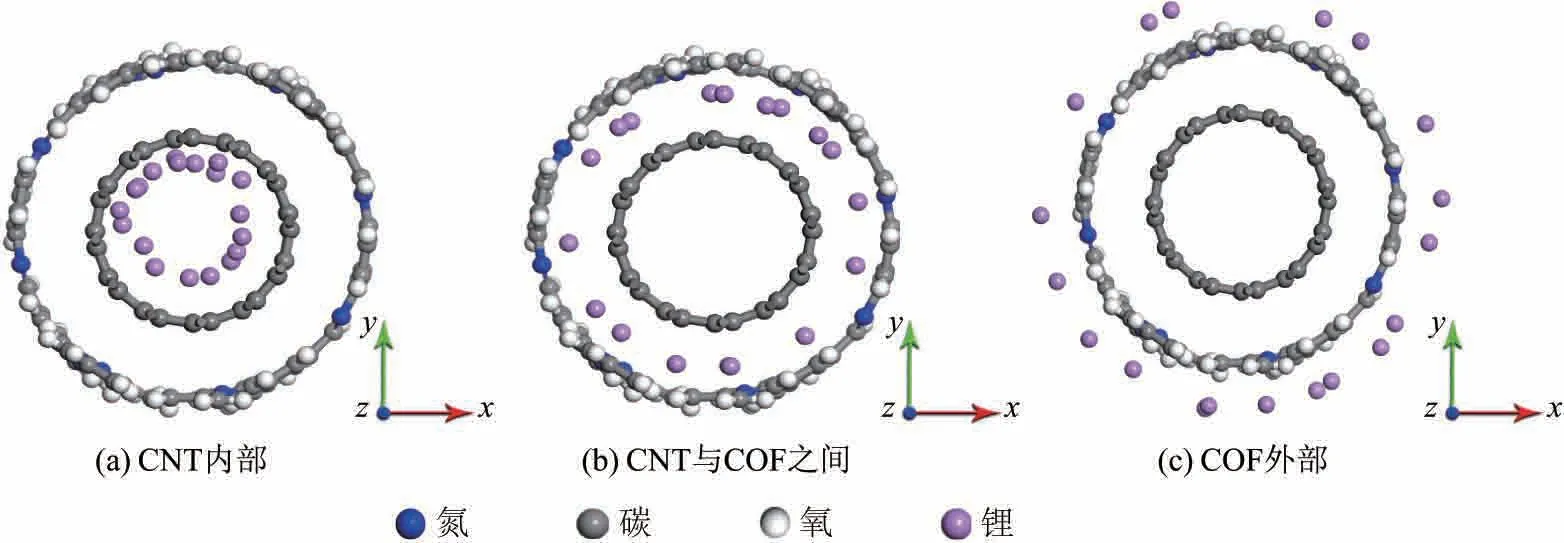
图8 Li+在COF@CNT中传输路径的位置示意图Fig.8 Position illustration for the transport trajectory of Li+in COF@CNT

图9 Li+在传输过程中的均方位移Fig.9 Mean square displacement of Li+during the transporting process
Li+的扩散系数D可通过Einstein公式计算得到,
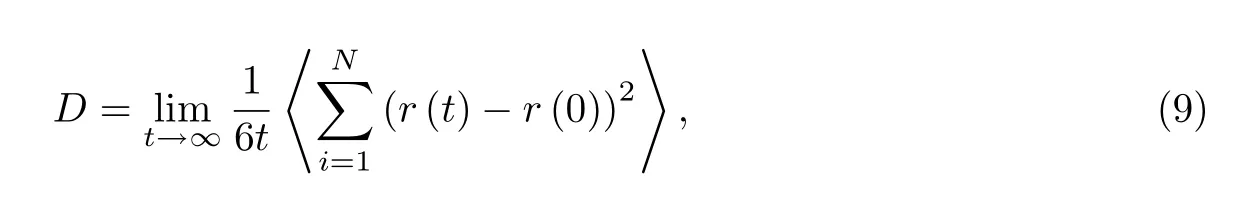
式中:t为模拟时长;r(0)与r(t)分别代表Li+在0与t时刻的位置矢量;〈·〉则表示系综平均,遍及所有在COF@CNT中传输的Li+.
Li+的电导率可通过Nernst-Einstein(NE)方程求得,

式中:NA为Avogadro常数;e是电荷数;KB是Boltzmann常数;T为体系温度.
表3为Li+在COF@CNT中的扩散系数及相应的电导率.显然,CNT内部的Λ最大,CNT与COF之间的Λ次之,且二者均高于Li+在单纯CNT中的电导率,而COF外部的Λ则最小.其主要原因在于,CNT内部为Li+传输的主要通道.与此同时,COF与CNT之间存在的π-π堆积效应又使CNT内外部的电子云密度发生显著变化,从而进一步提升了导电能力.而对于COF外部,由于Li+发生自由扩散的可能性显著增大,其沿CNT轴向进行有效传输的可能性自然减小,故Λ最小.
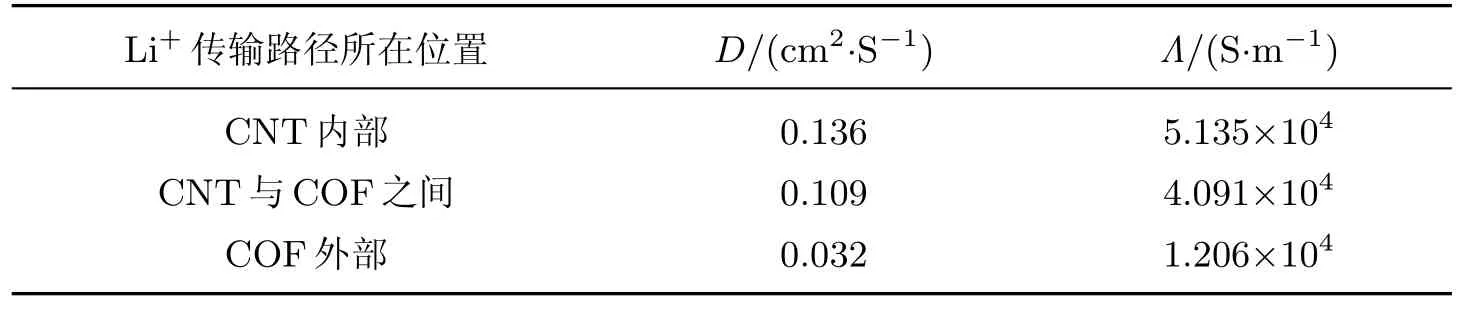
表3 Li+在COF@CNT中的扩散系数及相应的电导率Table 3 The diffusion coefficient and corresponding electronic conductivity of Li+in COF@CNT
3 结论
本工作针对COF@CNT复合材料中Li+的吸附与传输特性开展了分子模拟研究,明确了Li+的吸附位点与吸附顺序,得到了相应的吸附能,并观察到了COF@CNT的表观形貌变化.当达到饱和吸附状态时,COF@CNT的体积变化率仅为0.25,平均电压保持在2.00 V以上,而理论容量则高达1 402.47 mAh/g.此外,Li+在COF@CNT内部的电导率大于其在单纯CNT中电导率的实测值.研究结果可为此类物质的实际应用提供必要的理论帮助与指导.
致谢感谢上海大学高效能计算中心提供的支持!
猜你喜欢
储能科学与技术(2022年2期)2022-02-19
煤(2022年2期)2022-02-17
航空学报(2019年6期)2019-07-18
北京航空航天大学学报(2019年3期)2019-04-08
三联生活周刊(2017年48期)2017-11-25
科技知识动漫(2017年4期)2017-04-15
课堂内外·教师版(2017年3期)2017-04-13
食品工业科技(2014年13期)2014-12-16
储能科学与技术(2014年5期)2014-02-27
储能科学与技术(2014年5期)2014-02-27
