
STING介导的炎症在心血管疾病中的作用
2022-12-20 12:08聂晓妍王林涛何朝勇
药学研究 2022年11期
聂晓妍,王林涛,何朝勇
(中国药科大学药学院,江苏 南京 211198)
固有免疫反应作为机体识别、抵御病原体的第一道防线,产生大量免疫效应分子,维持机体内环境稳定。病毒及细菌等微生物侵入生物体产生的DNA、RNA、脂多糖及肽聚糖等成分被称为病原体相关分子模式(pathogen-associated molecular patterns,PAMPs)[1];机体自身创伤、应激后释放的DNA、高迁移率族蛋白1(high mobility group box 1,HMGB1)等物质被称为危险相关分子模式(damage-associated molecular patterns,DAMPs)。PAMPs和DAMPs均可被模式识别受体(pattern recognition receptors,PRRs)识别,DNA感受器作为胞质中重要的PRRs,识别胞质中游离的DNA,激活固有免疫反应。
环状鸟苷酸-腺苷酸合成酶(cyclic GMP-AMP synthase,cGAS)、DEAD-box RNA解旋酶41(DEAD-Box Helicase 41,DDX41)、干扰素诱导核蛋白16(interferon gamma-inducible protein 16,IFI16)等DNA感受器通过不同的方式激活干扰素基因刺激因子(STING)。cGAS通过合成2′3′环状鸟苷酸-腺苷酸(2′-3′-cyclic GMP-AMP,2′3′-cGAMP)激活STING,而DDX41及IFI16直接激活STING。DNA感受器作为DNA受体及STING的上游通路元件介导了STING的活化,STING是固有免疫反应中的关键接头蛋白。STING活化后通过内质网-内质网高尔基体中间体(reticulum-Golgi intermediate compartment,ERGIC)-高尔基体途径囊泡运输,激活下游TANK 结合激酶1(TANK-binding kinase 1,TBK1)、干扰素调节因子3(interferon regulatory factor 3,IRF3)及核转录因子-κB(nuclear factor-kappa B,NF-κB)等下游通路信号分子。IRF3及NF-κB可以上调干扰素(interferon,IFNs)、干扰素刺激基因(interferon stimulated genes,ISGs)及炎症因子水平,调控心血管疾病、自体免疫疾病、肿瘤等疾病的病理进程。现就STING介导的炎症及其在心血管疾病中的作用、STING抑制剂的发展情况进行综述。
1 STING介导的炎症反应
双链DNA(double-strand DNA,dsDNA)通过DNA感受器激活STING。正常情况下,DNA存在于细胞核、线粒体等膜隔开的区室中,同时细胞中脱氧核糖核酸酶(deoxyribonucleases,DNases)会使逃逸DNA失活。细胞核或线粒体通透性增加、DNases失活会导致胞质dsDNA异常堆积,通过DNA感受器过度激活STING。激活的STING通过内质网- ERGIC-高尔基体途径囊泡运输,运输过程中伴随着STING的构象改变及下游通路的激活。STING在内质网形成多聚物后通过ERGIC转运至高尔基体,在高尔基体,STING的Cys88及Cys91位点发生棕榈酰化,从而活化TBK1并磷酸化IRF3,STING也可以直接或间接激活NF-κB。STING-TBK1-IRF3或STING-NF-κB途径促进下游炎症因子表达增加,介导机体内炎症反应的发生。
1.1 STING信号通路 STING包含4个跨膜螺旋(TM1、TM2、TM3和TM4),N端细胞质配体结合域(LBD)和C端信号域(CTT)。非激活状态下,LBD在内质网的胞质侧形成二聚体,跨膜螺旋在STING二聚体中采用结构域交换结构,二聚体中的8个跨膜螺旋组织成两层:来自两个亚基的TM2和TM4形成中心层,外围被TM1和TM3包围。跨膜区域和LBD相互作用,形成完整的域交换二聚体。TM4和LBD中的首个螺旋之间的连接子(LBDα1)之间存在一个两亲性螺旋(以下称为连接器),连接器通过连接器环连接到LBDα1形成连接器- LBDα1元件,静息状态二聚体中两个连接器-LBDα1元件形成右手交叉结构。cGAMP 与STING结合后,STING发生蛋白结构重排,促使STING通过并排堆积形成寡聚体[13]。
内质网STING通过ERGIC转运至高尔基体,在高尔基体中发生Cys88与Cys91位点棕榈酰化进一步促进STING招募并活化TBK1及IRF3 。近期发现,高尔基体内合成的硫酸化糖胺聚糖(sGAGs)作为STING共配体介导STING在高尔基体的活化,sGAGs与STING的高尔基体内侧极性氨基酸结合,像铰链一样引发STING多聚化[14]。STING在高尔基体的进一步寡聚使其具备了招募TBK1及IRF3的条件。CTT对于STING激活TBK1及IRF3是必需的,CTT中存在一个保守的共识基序 (pLxIS;p是亲水性残基,x是任何残基),其在人STING的Ser366位点(小鼠位点在Ser365)磷酸化由TBK1介导并招募活化IRF3。另外,STING在内质网移位过程中激活NF-κB抑制因子激酶(IκB kinase,IKK),从而磷酸化IκB,使其通过泛素-蛋白酶体途径降解,释放出游离的NF-κB[16]。除此之外,胞质肿瘤坏死因子受体相关因子6(TNF receptor associated factor 6,TRAF6)E3泛素连接酶介导STING连接K63多泛素链,通过转化生长因子β激活激酶1(TGF-β activated kinase-1,TAK1)、TAK1结合蛋白2/3(TAK1 binding protein2/3,TAB2/3)和IKK,激活NF-κB。尽管以上结果表明STING直接激活NF-κB引起炎症反应,但也有文献证明STING通过TBK1激活NF-κB[17]。此信号传递过程复杂,目前仍有待进一步解决。
此外,STING还可以在某些特殊情况下激活,如STING蛋白结构中的特殊位点发生突变、NOD样受体家族含CARD结构蛋白3(NOD-like receptor family CARD domain containing 3,NLRC3)与DNA的结合、尼曼匹克蛋白1(Niemann-Pick disease type C1,NPC1)的缺失等。N154S、V147L、V155M等STING的突变类型可以引起STING自发活化。NLRC3与STING结合使其处于抑制状态,DNA存在的情况下,NLRC3释放并激活STING。NPC1是STING进入溶酶体的接头蛋白,NPC1缺失抑制STING进入溶酶体降解,激活STING[22]。
因此,STING作为胞内重要的接头蛋白,在多种因素刺激下均有作用,研究其下游发挥的不同作用将有利于剖析STING介导的生理功能与炎症发生。
1.2 STING-TBK1-IRF3介导的炎症反应 STING-TBK1-IRF3途径的激活可能需要内质网膜蛋白胰岛素诱导基因1(insulin induced gene-1,INSIG1)、SREBP裂解激活蛋白(SREBP-cleavage activating protein,SCAP)的辅助。INSIG1介导E3泛素连接酶自分泌运动因子受体(autocrine motility factor receptor,AMFR)对STING的K27泛素化,这有利于TBK1的招募[23]。STING通过SCAP 与IRF3连接,促进STING磷酸化IRF3。磷酸化的IRF3形成二聚体进入细胞核与特定基因启动子结合促使IFNs、ISGs及炎症因子表达上调(见图1)。
NPC1缺失引起小鼠尼曼匹克病C型表型,表现为小脑中浦肯野细胞丢失、小胶质细胞激活,炎症反应发生。NPC1敲除小鼠脑组织及巨噬细胞中的STING激活,促使趋化因子配体1(C-X-C motif chemokine ligand,CXCL1)、CXCL10、趋化因子2(C-C chemokine ligand 2,CCL2)、CCL5等炎症趋化因子及干扰素诱导的三角形四肽重复蛋白1(interferon-induced protein with tetratricopeptide repeats 1,IFIT1)、IFIT2、IFIT3等ISGs的mRNA水平升高,血清中巨噬细胞炎性蛋白-1α(macrophage inflammatory protein -1α,MIP-1α)、MIP-1β、IL-13、嗜酸性粒细胞趋化因子(eotaxin)表达增加,STING或IRF3敲除后上述趋化因子、ISGs的mRNA水平及炎症因子表达降低,小脑中浦肯野细胞丢失减少,小胶质细胞激活减少[22]。游离脂肪酸引起人正常肝脏细胞中STING-IRF3通路激活,肝细胞中IL-6、IL-1β等炎症因子升高,这种升高可以通过敲除STING或IRF3逆转。二氧化硅颗粒能引起肺部炎症及肺纤维化,导致硅肺病的发生。硅肺病患者肺组织中STING-TBK1-IRF3途径激活,CXCL10、IFNβ表达升高,此外在硅肺病小鼠模型中也观察到了TNF-α、IL-1β表达增加。上述通过STING-IRF3通路介导升高的炎症因子促进了机体内炎症反应的发生,造成了机体不同程度的损伤。
1.3 STING-NF-κB介导的炎症反应 STING在高尔基体上激活NF-κB抑制因子(inhibitor of NF-κB,IκB)激酶(IκB kinase,IKK)[16],磷酸化转录因子IκB,使其通过泛素-蛋白酶体途径降解,释放出游离的NF-κB。角质细胞核DNA损伤信号传到胞质肿瘤坏死因子受体相关因子6(TNF receptor associated factor 6,TRAF6)激活STING。在此过程中TRAF6作为E3泛素连接酶介导STING连接K63多泛素链,K63多泛素链组装转化生长因子β激活激酶1(TGF-β activated kinase-1,TAK1)、TAK1结合蛋白2/3(TAK1 binding protein 2/3,TAB2/3)和IKK,激活NF-κB,上调炎症水平。尽管以上结果表明STING直接激活NF-κB引起炎症反应,但也有文献证明STING通过TRAF6-TBK1轴激活NF-κB[17]。综上,STING-NF-κB是否通过TBK1介导还有待进一步阐明。NF-κB活化后进入细胞核诱导如肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、白介素-1β(interleukin-1β,IL-1β)及白介素-6(interleukin- 6,IL-6)等炎症因子表达上调(见图1)。
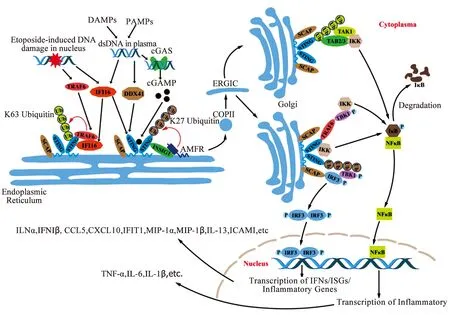
图1 STING活化后通过TBK1-IRF3及NF-κB通路激活炎症反应
高脂喂养诱导小鼠发生非酒精性脂肪性肝炎,该模型中肝脏枯否细胞的STING激活,NF-κB 表达水平上调,IL-6、IL-1β、TNF-α等炎症因子水平升高,这种升高在STING敲除的小鼠中被抑制,并可被NF-κB 抑制剂BAY11-7082进一步逆转[26]。近期在急性肾损伤与慢性肾损伤的小鼠模型中发现STING-NF-κB通路的激活。顺铂诱导小鼠发生急性肾损伤,该模型中肾小管上皮细胞线粒体损伤,线粒体DNA泄漏到胞质激活cGAS,活化STING-NF-κB途径,上调IL-6、细胞间黏附分子-1(intercellular cell adhesion molecule-1,ICAM1)、CXCL10、粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony-stimulating factor,GM-CSF)等炎症因子及趋化因子水平。肾小管细胞特异性敲除线粒体转录因子A(mitochondrial transcription factor A,TFAM)的小鼠作为慢性肾损伤的模型,发现该模型小鼠肾小管内的线粒体大量释放激活STING-NF-κB通路,IL-6、IL-1β、TNF-α、CCL2等上调,导致肾脏慢性炎症。STING-NF-κB途径激活IL-6及TNF-α等经典炎症因子,上调机体内的炎症反应。
2 STING介导的炎症促进心血管疾病病理进程
炎症与心血管疾病的发生密切相关,近年来STING介导的炎症反应在心血管疾病研究中取得一定进展。在心血管系统中,STING主要表达在免疫细胞、血管内皮细胞、血管平滑肌细胞(vascular smooth musle cells,VSMC)、心肌细胞及心肌成纤维细胞。细胞死亡或线粒体受损导致dsDNA释放,激活DNA感受器,活化STING通路,介导炎症因子转录上调,这可能是心血管疾病中炎症发生的重要机制。
2.1 内皮损伤 内皮细胞作为血管炎症中重要的驱动细胞,其在动脉粥样硬化起始阶段或慢性代谢性疾病相关的炎症反应起着重要作用。肥胖致使外周循环中游离脂肪酸含量增加,促使血管内皮细胞线粒体DNA泄漏到胞质激活cGAS,活化STING-IRF3途径,上调ICAM-1,导致血管内皮炎症反应的发生。血管通透性改变是脓毒症致死过程中重要环节,LPS引起血管内皮细胞焦亡发生,增加的胃泌素D(gasdermin D,GSDMD)活化促使线粒体DNA释放,从而激活cGAS-STING,抑制内皮细胞增殖,最终导致血管内皮通透性改变。上述充分表明STING参与着内皮细胞炎症反应的发生与发展。
2.2 动脉粥样硬化 动脉粥样硬化(atherosclerosis,AS)是一种慢性炎症疾病,是临床心血管事件的主因。巨噬细胞和VSMC参与了AS的炎症发生过程。研究发现在高脂饮食喂养ApoE敲除小鼠中巨噬细胞特异性表达STING引发血管炎症,这可能是由TDP43诱导线粒体DNA释放介导的[31]。AS的血管斑块中,VSMC会从收缩型转向合成型,分泌细胞外基质(extracellular cell matrix,ECM)构成纤维帽,增加斑块稳定性,降低心血管事件的发生。慢性肾病(chronic kidney diseases,CKD)促使VSMC过早衰老,并使其以自分泌/旁分泌的方式进行表型转换,导致纤维帽中血管平滑肌细胞丢失,纤维帽变薄,斑块破裂易感性增加,加速了AS的斑块破裂进程。在上述过程中,CKD诱导的氧化应激导致VSMC线粒体损伤,线粒体通透性转换孔(mitochondrial permeability transition pore,MPTP)开放,线粒体DNA释放入胞浆并通过cGAS-STING通路上调Ⅰ型IFNs,激活Janus激酶(Janus kinase,JAKs)-转录激活因子1(signal transducer and activator of transcription 1,STAT1)信号通路,引发炎症反应 。以上研究证明,线粒体DNA通过STING-IRF3途径介导VSMC的炎症反应,促进As发展。
2.3 主动脉剥离 主动脉瘤及主动脉夹层(aortic aneurysm and dissection,AAD)表现为进行性VSMC丢失及ECM碎裂、耗竭,主动脉瘤、主动脉夹层形成,最终发展为主动脉破裂。Wei等在人和小鼠的AAD样本中发现,主动脉VSMC的dsDNA泄漏至胞质激活STING,引发细胞坏死导致dsDNA释放入血管壁,招募活化巨噬细胞。dsDNA在巨噬细胞中活化STING-TBK1-IRF3通路,IRF3与基质金属蛋白酶9(matrix metalloproteinase-9,MMP9)启动子结合上调,MMP9表达增加并释放入血管壁,破坏血管弹力板,促进AAD进程。
2.4 心肌梗死 实验表明,过度活跃的炎症信号会导致心肌梗死后左心室扩张增加,收缩功能障碍。心肌梗死后,骨髓及肾脏来源的单核细胞被招募至心脏,初期吞噬细胞碎片分化为促炎的M1型巨噬细胞,单核细胞对凋亡细胞的有效清除可能促使巨噬细胞偏向修复的M2型转化。心肌梗死后心肌组织的修复对于恢复心肌功能至关重要,抑制STING通路促使心肌梗死后招募的巨噬细胞偏向修复的M2型转化。CXCL10与巨噬细胞的M1样极化相关,在心肌梗死模型小鼠心脏的巨噬细胞中高表达;CD163、IL-10和CCL17是巨噬细胞的M2型标记物,在cGAS敲除的心肌梗死模型小鼠的心脏中表达增加。心肌梗死的小鼠模型中STING和cGAS表达水平升高,STING-IRF3通路活化,IFNβ、CXCL10、IRF7等因子表达上调,STING敲除抑制了上述因子的表达。Lai等[39]证明了小鼠心肌缺血损伤后,心肌细胞释放DNA及HMGB1,再灌注时二者进入循环激活炎症反应,引起缺血再灌注损伤。在心肌缺血再灌注小鼠中使用STING抗体阻断Ⅰ型IFNs信号通路,可显著减小梗死面积。以上研究证明免疫细胞中cGAS监测到心肌梗死释放的DAMPs,并通过STING-IRF3途径介导了心肌梗死中炎症的发生。
2.5 系统性炎症导致的心肌损伤 STING参与介导了由二手烟、系统性红斑狼疮(systemic lupus erythematosus,SLE)及脓毒症等危险因素引起的系统性炎症,导致心肌损伤。侧流烟雾在实验中用于模拟二手烟吸入,引起小鼠心肌细胞线粒体损伤,线粒体DNA释放入胞质,激活STING通路,诱导TNF-α和IL-1β表达水平上调,导致心脏结构、功能异常,敲除单个Beclin1等位基因加重了侧流烟雾引起的炎症反应及心功能障碍。这是因为Beclin1参与细胞自噬小体的组装,清除细胞内大量产生的线粒体DNA,有助于抑制dsDNA诱导的STING过度磷酸化,避免炎症反应过度发生。生理状况下, 3′-核酸修复外切酶1 (three prime repair exonuclease 1,TREX1)清除胞质DNA,防止内源性DNA积聚,TREX1突变失活会导致系统性红斑狼疮(systemic lupus erythematosus,SLE)的发生[41]。SLE是一种自身免疫疾病,相较正常人SLE患者更易患心血管疾病,1/3的SLE死亡是心血管事件引起的[42]。TREX1基因敲除小鼠通过激活STING-IRF3通路产生高水平IFNs,导致心肌炎、血管炎等疾病发生,敲除cGAS可以遏制上述炎症反应。LPS诱导小鼠脓毒性心肌病模型,表现为心功能下降、炎症水平升高,小鼠的心脏射血分数及短轴缩短率在STING敲除后明显改善,炎症水平也明显降低。脓毒性心肌病模型小鼠的发病机制是STING在LPS刺激下活化NOD样受体热蛋白结构域相关蛋白3(NOD-like receptor protein 3,NLRP3),NLRP3在胞质中活化半胱天冬酶-1(cysteinyl aspartate specific proteinase 1,caspase-1),切割IL-1β及IL-18,升高炎症水平[44]。以上研究证实,上述危险因素引起的系统性炎症中,STING介导了心肌组织炎症反应的发生,造成心肌损伤。
2.6 化疗药物导致的心肌损伤 顺铂是广谱化疗药物,临床上发现顺铂可以造成心肌损伤[45]。我们的前期研究发现顺铂诱导心肌损伤的小鼠模型中TNF-α、IL-6等炎症因子表达上调,然而,敲除Sting基因可有效抑制顺铂引起的心肌炎症因子表达与心功能不全。在诸多临床化疗药物治疗患者中观察到长期的化疗治疗经历促使患者在多年后表现出心功能不全,STING介导的炎症反应在化疗药物诱导心肌损伤中的作用值得获取更多的关注。
2.7 心力衰竭 心力衰竭是心血管疾病的终末期临床综合征,表现为心脏收缩或舒张功能障碍,射血功能受损。扩张型和肥厚型心肌病的人类样本中发现STING、IFNα和IFNβ水平升高。用于模拟心力衰竭的主动脉缩窄(transverse aortic constriction,TAC)小鼠模型表现为心肌肥厚、心功能不全和心肌纤维化,STING、IFNα和IFNβ的表达增加,STING敲除的TAC小鼠心功能明显改善[46]。TAC术后3 d,小鼠心肌组织中IFNs、CXCL10、IFIT3和ISG15的表达水平显著升高,腺相关病毒9(adeno-associated virus,AAV9)沉默cGAS可显著减少小鼠心脏左室重构和纤维化[47]。上述研究证实了STING引起的炎症反应促进了心力衰竭的病理进程。
2.8 STING相关的婴儿期发病血管病变 STING相关的婴儿期发病血管病变(STING-associated vasculopathy with onset in infancy,SAVI)是单基因突变疾病,表现为全身炎症,严重的皮肤血管病变、间质性肺病及反复细菌感染。该突变促使STING在无需配体的情况下从内质网运往核周小泡中聚集活化,激活下游炎症相关通路,上调干扰素表达,促使炎症发生,其中STING突变体类型分为遗传型和自发型,自发型突变较遗传型发病早,程度也更为严重,如V155M。N154S、V155M是SAVI患者中常见的突变表型,相较于N153S突变类型小鼠,V154M小鼠具备更强的疾病表型,这或许解释了SAVI患者群体中发病程度的差异性;IRF3及NF-κB通路同时参与了N153S、V154M小鼠中STING突变的下游机制[49]。临床调查表明,除却V147L、V147M及跨膜区中S102P等少数突变位点,大多SAVI患者的STING突变位点如N154S、V155M、V155E、R281Q、R284S、G207E等,位于STING的cGAMP结合结构域。SAVI的发生与上述STING突变引起的多聚化相关,其中V147L、N154S、V155M突变发生于STING的C148位点附近,多个STING蛋白的C148位点之间形成二硫键促使STING多聚化进而激活,实验表明V147L、N154S的作用依赖于C148[51]。上述研究表明STING功能获得性突变引起的炎症反应促进了SAVI发生。
3 STING抑制剂
上述研究证实,STING介导的炎症反应促进了心血管疾病的发展。因此靶向抑制STING,缓解炎症反应过度发生,为心血管疾病的治疗提供了一个新思路。现有STING抑制剂的开发主要针对STING的配体结合口袋及棕榈酰化位点靶点进行计算机辅助设计,通过高通量筛选筛选出候选分子,并验证候选分子对鼠源或人源STING的抑制效率。以配体结合口袋为靶点的抑制剂与STING的内源性配体cGAMP结合,抑制cGAMP对STING的激活作用,这类抑制剂主要包括SN-011、天然产物环肽Astin C、四氢异喹啉类(化合物1和化合物18)等。STING 的Cys88和Cys91位点发生棕榈酰化对于其活化过程中形成多聚复合物、招募下游信号通路分子是必要的。仅对Cys91位点有抑制作用的抑制剂包括硝基呋喃类(C176、C178、C170和C171)、H151及丙烯酰胺类(BPK-21和BPK-25),硝基脂肪酸类(nitro-fatty acids,NO2-Fas;NO2-cLA,NO2-OA)则对两个棕榈酰化位点均有抑制作用。研究表明STING抑制剂缓解了小鼠As的发展,提示STING抑制剂有望成为治疗心血管疾病的药物。研究证实以上抑制剂对鼠源STING有抑制效果,目前STING抑制剂对心血管疾病的治疗作用处于临床前研究阶段,其在临床心血管疾病中的作用尚需研究验证。
4 结语与展望
研究证实STING介导的炎症对心血管疾病有着促进作用。心血管疾病中,免疫细胞、血管内皮细胞、VSMC及心肌细胞等发生线粒体损伤或细胞死亡,导致线粒体或细胞核DNA泄漏到胞质中。胞质中DNA感受器在dsDNA的刺激下,通过STING-TBK1-IRF3上调IFNs、ISGs及ICAM-1、MIP-1β、IL-13、IL-1β等炎症因子水平,介导了肥胖中内皮损伤、CKD加剧的As中VSMC炎症,加重了AAD、心肌梗死及心力衰竭;或者通过STING-NF-κB途径增加IL-6、IL-1β、TNF-α等炎症因子表达,可能介导了顺铂引起的心肌损伤。在心血管系统炎症反应发生的过程中,STING起到了接头蛋白的作用,将细胞DNA感知与炎症反应联系起来。因此,靶向抑制STING为治疗心血管疾病提供了一个新思路。
前述STING抑制剂能否成药取决于其对人源STING的靶向性及安全性。SN-011、C170、C171及NO2-FAs、H-151对鼠源和人源STING均有抑制作用,但H151毒性较大且对STING特异性不高,SN-011的毒性相对较低但尚未经临床实验证实其有效性与安全性。目前CXA-10作为安全且耐受性良好的NO2-Fas已进入Ⅱ期临床试验(NCT04125745、NCT04053543、NCT03449524),目的是探究其对肺动脉高压的治疗效果。除STING抑制剂外, RU.521、G150、化合物S3、A151、CU-76、阿司匹林等cGAS抑制剂及氨来占诺、化合物 II、BX795、Domainex等TBK1抑制剂也可以抑制STING通路介导的炎症反应。其中氨来占诺是临床上市药物且减缓了小鼠AAD的进程,阿司匹林用于治疗COVID-19患者体内STING通路活化引起的血栓性凝血功能障碍。综上,STING通路抑制剂有望成为临床心血管疾病的治疗药物。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
材料与冶金学报(2022年2期)2022-08-10
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
天津医药(2016年9期)2016-10-20
中国老年学杂志(2015年16期)2015-03-05
癌变·畸变·突变(2015年4期)2015-02-27
癌变·畸变·突变(2014年1期)2014-03-01
