
基于单倍型肉牛屠宰性状全基因组关联分析研究
2023-01-03 02:28李宏伟徐凌洋王泽昭蔡文涛张路培高会江李俊雅
畜牧兽医学报 2022年12期
李宏伟,徐凌洋,王泽昭,蔡文涛,朱 波,陈 燕,高 雪,张路培,高会江,李俊雅
(中国农业科学院北京畜牧兽医研究所 牛遗传育种创新团队,北京 100193)
随着高通量测序技术的发展和大量SNP标记的出现,全基因组关联分析逐渐成为定位复杂性状遗传变异或基因的常用方法。该方法于1996年由Risch和Merikangas[1]首次提出。近二十年来,科学家们在人类、牲畜和作物中开展了大量基于连锁不平衡的GWAS研究工作[2-9]。GWAS研究有两个具体目标,一是要确定与目标性状关联的QTLs或者基因,其次是探究复杂性状的遗传结构[10]。全基因组关联分析开始广泛应用更高密度的芯片及全基因组测序数据开展基因定位研究,大大提高了GWAS的检测效率。伴随GWAS研究的深入,GWAS新的统计模型和算法不断发展,用以提高位点定位的精准性。2006年,Yu等[11]首次提出基于固定效应和随机效应的混合模型。随后,针对GWAS研究中的计算速度和假阳性等问题陆续提出了优化模型和方法。这些方法主要分为两类,一类是精确算法,另一类是近似算法。精确算法如GEMMA和fast-LMM等针对每个标记重新拟合模型,并准确计算每个标记的效应值,然而,此类方法需要较长的计算时间[12-13]。相比之下,近似算法如EMMAX和GRAMMAR的计算速度非常快。单倍型是指在同一染色体上进行共同遗传的多个基因座上等位基因的组合。多个等位基因之间存在一定的连锁不平衡关系。单倍型标记与数量性状基因座之间具有较强的LD关系,在基因定位、因果突变鉴定及QTL连锁分析方面具有较高的统计效力。然而,目前大部分畜禽基因组研究工作都是基于SNP标记,利用单倍型标记的研究较少。有研究表明,基于单倍型的全基因组关联分析能够检测到基于SNP检测不到的位点[14-15]。单倍型可以捕获SNP之间的局部上位性效应,因此可以提高解析复杂性状的能力和准确性[16-17]。在人类的相关研究中已经揭示了利用单倍型开展GWAS研究的诸多优点:首先,与SNP标记相比,单倍型标记提供了更多的多态性信息[18];其次,单倍型标记可以用来判断两个等位基因在血统上的一致性[19];另外,利用单倍型标记可以捕获更多的物种进化信息[20]。目前,单倍型已日益成为全基因组关联分析中重要的遗传标记[21-25]。
本研究基于中国农业科学院北京畜牧兽医研究所构建的华西牛资源群体,分别利用SNP和单倍型标记对宰前活重(LW)和屠宰率(DP)开展全基因组关联分析,定位目标性状的分子标记及候选基因。
1 材料与方法
1.1 试验群体
本试验的研究群体为中国农业科学院北京畜牧兽医研究所牛遗传育种团队构建的华西牛资源群体,选取该群体于2008—2021年间屠宰的共计1 478个个体进行研究,其中公牛1 333头,母牛145头。所有个体的表型数据从出生时开始收集整理,包括各个生长发育的阶段的表型数据,犊牛断奶后,公牛犊转移至指定的肉牛育肥场进行育肥,按照相同的饲养方式进行统一管理,并于24月龄左右进行屠宰,按照国家标准GB/T 27643-2011进行屠宰、胴体、分割等性状的测定和收集。
1.2 试验方法
1.2.1 表型性状测定及数据预处理 在1 478头华西牛群体中,公牛1 333头,母牛145头,选择宰前活重(LW)和屠宰率(DP)性状进行研究。具体的测定计算方法:宰前活重为肉牛屠宰前禁水禁食12 h以上的体重;屠宰率是肉牛胴体重与宰前活重的比值。
首先对原始数据进行预处理,剔除各个性状平均值加减3倍标准差之外的异常数据,然后对各性状进行描述性统计分析,并进行表型校正。本研究根据资源群体数据的实际情况,考虑的固定效应为性别、出生年、育肥场、屠宰场和所在群体等,协变量包含育肥天数、进场重和屠宰日龄等。利用R语言中的GLM函数对影响原始表型的各个因素进行显著性检验和校正,其模型如下:
y=u+Sex+Year+F1+F2+Pop+day1+enter_wt+day2+e
其中,y为表型值,u为总体均值,Sex为性别效应,Year为出生年效应,F1为育肥场效应,F2为屠宰场效应,Pop为所在群体效应,day1为育肥天数效应,enter_wt为进场重效应,day2为屠宰日龄效应,e为随机残差。
1.2.2 遗传参数估计 本研究利用ASReml (v4.1)通过个体动物模型对目标性状的遗传力进行了估计[26]。具体模型如下:
y=1nμ+Za+e

1.2.3 基因型数据处理 利用PLINK软件(v1.07)[27]对1 478个个体的770K高密度芯片数据进行质量控制,质控后利用BEAGLE(v5.1)[28]对芯片数据中缺失位点进行填充,具体质控方法:首先,去除性染色体和质粒上的SNPs位点,仅保留常染色体上的位点。剔除染色体位置信息缺失的SNPs位点;剔除最小等位基因频率小于0.01的SNPs位点;剔除缺失率大于0.05的SNPs位点;剔除哈代-温伯格平衡检验P<10-6的SNPs位点;剔除样本SNP缺失率大于0.05的个体。
1.2.4 基因型定相及单倍型构建 本研究利用BEAGLE (v5.1)软件的默认参数对质控后的SNP数据进行定相(phase)。分别采用基于LD水平和基于连续固定的SNP个数的方式进行单倍型的构建,具体过程如下。
1.2.4.1 基于LD单倍型构建:利用相邻两个SNPs之间的连锁不平衡程度(LD)进行单倍型的划分。若某一区域彼此相邻两个SNPs之间的LD大于某一个阈值(r2),则将这部分连续SNPs划分为一个单倍型块。这种方法确保在物理距离尽可能近的情况下,具有一定LD关系的SNPs可以划分到一个单倍型区域中。r2的计算公式如下:
其中,D=pA1B1pA2B2-pA1B2pA2B1,D是两个双等位基因之间的连锁不平衡程度,pA1、pA2、pB1和pB2是4个等位基因(A1、A2、B1和B2)的频率,pA1B1、pA2B2、pA1B2和pA2B1是4个基因型频率(A1B1、A2B2、A1B2和A2B1)[29-30]。本研究选择LD阈值水平为r2>0.3进行单倍型(0.3LD)的构建。
1.2.4.2 基于固定SNP个数进行单倍型构建:本研究将整个基因组按常染色体进行拆分(共29条),在每条染色体上,依据SNP的排列顺序,设置每5个连续的SNPs划分为一个单倍型块(5_SNP_HAP)。若染色体末端SNP个数不足5个,将染色体末端最后剩余的所有SNP作为一个单倍型块。在单倍型块内部按照剂量可加效应,依据单倍型等位基因的重复个数进行编码(0、1、2)。
为了排除低频单倍型等位基因对GWAS结果的影响,对单倍型等位基因进行质量控制,去除单倍型等位基因频率小于0.01的单倍型等位基因。
1.2.5 单倍型效应编码 在划分单倍型块的基础上构建单倍型矩阵。具体编码规则是利用剂量可加效应,依据单倍型等位基因重复个数的方法进行编码[31],两个连续SNPs构建一个单倍型块的剂量可加编码规则如表1所示。
1.2.6 群体遗传结构分析 在全基因组关联分析时,为了减少假阳性位点的出现,在分析之前需要对群体分层进行校正。本研究利用高密度SNP芯片数据对华西牛群体做PCA分析,判断群体中是否存在群体分层现象。
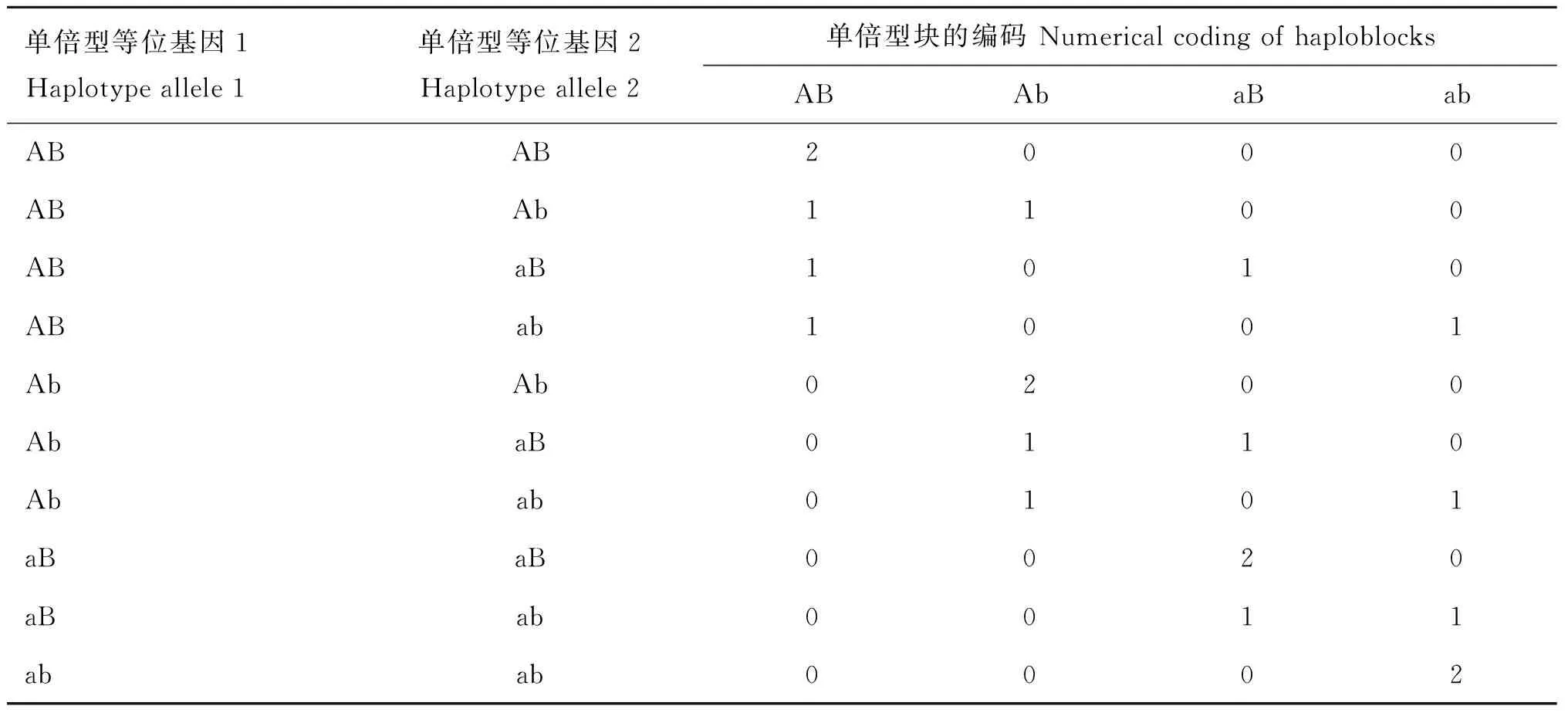
表1 两个连续SNPs构建一个单倍型块的剂量可加编码
1.2.7 全基因组关联分析
1.2.7.1 基于SNP的全基因组关联分析:利用GCTA软件(v1.93)[32]中的混合线性模型(mixed linear model,MLM)对目标性状进行GWAS研究。计算模型如下:
y=Xβ+Ws+Zμ+e
其中,y是表型值向量;X是固定效应向量的关联矩阵,β是固定效应向量;W是SNP基因型指示变量,aa、Aa和AA三种不同基因型分别用0、1、2数字编码,s为SNP效应向量;Z为多基因效应向量的设计矩阵,μ~(0,Kσ2)是微效多基因效应向量,K为基因组亲缘关系矩阵;e~N(0,Iσ2)表示随机残差。
1.2.7.2 基于单倍型的全基因组关联分析:利用连续5个SNPs构建的单倍型的全基因组关联分析模型如下:
y=Xβ+W′s′+Z′μ′+e
其中y是表型值向量;X为固定效应向量的关联矩阵,β为固定效应向量;W′是单倍型的指示变量,依据单倍型等位的重复个数(0、1、2)进行编码,s′为单倍型等位基因效应向量;Z′为多基因效应向量的设计矩阵,μ′~(0,K′σ2)为微效多基因效应向量,K′为单倍型等位基因计算的基因组亲缘关系矩阵;e~N(0,Iσ2)表示随机残差。
由于基于LD进行单倍型构建时将整个基因组划分为两部分,一部分为具有LD的区域,利用单倍型进行重编码,剩余的位于单倍型块外部的SNP依旧利用原来的方式进行编码。因此,利用LD单倍型全基因组关联分析的模型如下:
y=Xβ+W″s″+Z″μ″+e
其中,y是表型值向量;X为固定效应向量的关联矩阵,β为固定效应向量;W″是单倍型和SNP合集的指示变量,其中单倍型部分依据单倍型等位的重复个数(0、1、2)进行编码,s″为SNP与单倍型等位基因效应向量;Z″为多基因效应向量的关联矩阵,μ″~(0,K″σ2)为微效多基因效应向量,K″为单倍型等位基因和单倍型外部SNP计算的基因组亲缘关系矩阵;e~N(0,Iσ2)表示随机残差。
采用wald检验法对得到的SNP或单倍型等位基因进行显著性检验,利用bonferroni多重检验将SNP标记的显著水平阈值设置为1/SNP数[33],单倍型等位基因显著性水平阈值设置为1/单倍型等位基因数。
1.2.8 候选基因筛选 根据设定的显著性阈值筛选与目标性状显著关联的SNPs及单倍型块,并通过生物数据库ensembl中的BioMart模块将筛选到的显著SNPs与牛的参考基因组(Bos_taurus.UMD3.1)进行比对。然后利用SNPs或单倍型块的物理位置寻找距离SNP最近的基因。且认为该基因是与目标性状显著相关的候选基因,利用NCBI网站的Gene数据库检索相关候选基因的生物学功能。
2 结 果
2.1 芯片数据质控与表型基本统计量
经过基因型数据质控及表型预处理后宰前活重和屠宰率性状分别剩余1 387及1 366个个体,共672 060个SNPs位点的基因型数据用于下一步分析。各性状表型的基本统计量,包括表型数据的记录数、平均值及标准差、最大值、最小值和变异系数如表2所示。图1展示了性状的表型频率分布情况。研究结果表明表型数据基本服从正态分布,可用于下一步研究。

表2 性状基本统计量
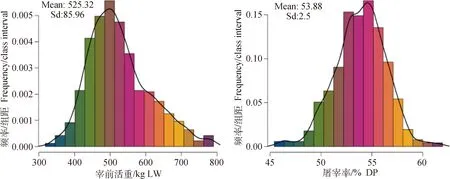
图1 宰前活重和屠宰率性状的表型频率分布图Fig.1 Phenotypic frequency distribution of LW and DP
2.2 固定效应及协变量显著性
对性别、出生年、育肥天数、进场重、育肥场、屠宰场、所在群体和屠宰日龄等固定效应和协变量进行显著性检验,使用GLM函数对各性状进行表型校正,检验结果如表3所示。
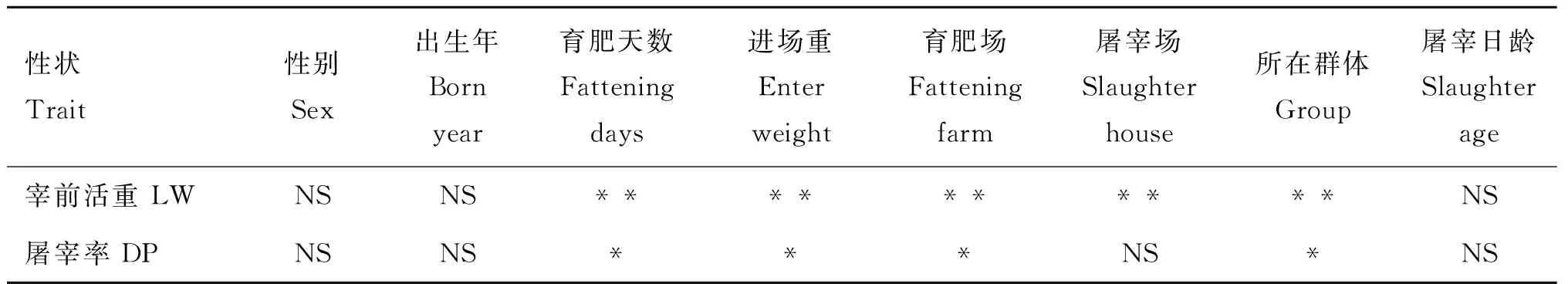
表3 各固定效应及协变量显著性结果
结果显示,各个性状中,性别、出生年和屠宰日龄对宰前活重和屠宰率的影响不显著(P>0.05),育肥天数、进场重、育肥场、屠宰场和所在群体对宰前活重的影响极显著(P<0.01),育肥天数、进场重、育肥场和所在群体对屠宰率影响显著(P<0.05)。
2.3 表型遗传参数估计
利用ASReml(v3.0)软件[34]中的个体动物模型对各性状的遗传参数进行估计。各性状加性遗传方差、残差方差和遗传力如表4所示。宰前活重和屠宰率的遗传力分别为0.40和0.22。其中,宰前活重为高遗传力性状,屠宰率为中等遗传力性状。
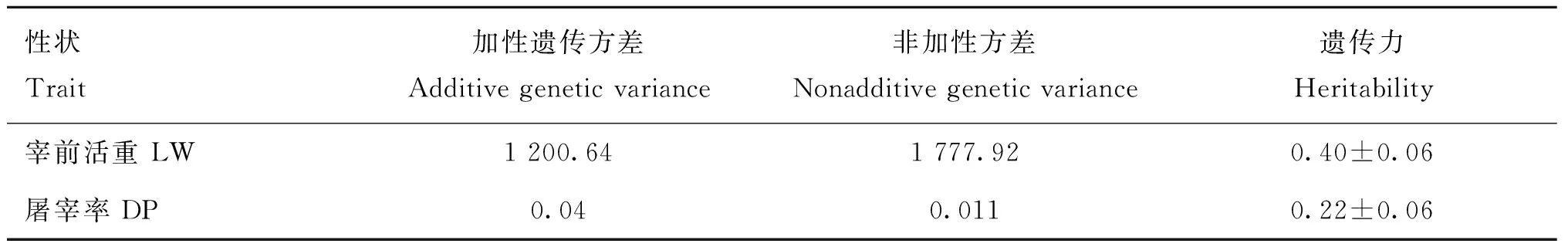
表4 性状遗传参数估计
2.4 群体遗传结构
以第一、二主成分聚类的群体结构如图2所示。群体中的个体大致聚成5类,表明群体中存在一定程度的群体分层现象。本研究根据前期研究的经验,选取前5个主成分作为协变量放入GWAS分析模型以校正群体分层效应对关联分析的影响。
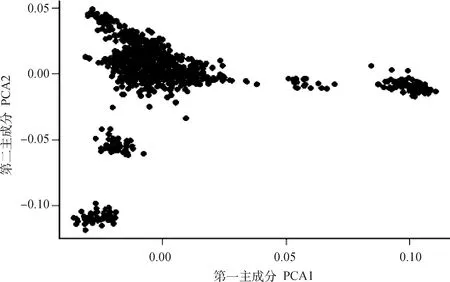
图2 主成分分析绘制种群结构图Fig.2 Population structure identified by principal components analysis
2.5 全基因组关联分析结果
图3和图4分别展示了基于SNP、0.3LD单倍型和5_SNP_HAP单倍型标记对宰前活重和屠宰率等性状的GWAS分析的Manhattan和QQ图。QQ图结果显示,不论SNP标记还是单倍型标记,SNP显著性分布的理论值与观察值的基本吻合,表明本研究选取的固定效应和协变量较好的校正了群体结构和其他混杂因素,模型选择合理。以bonferroni法矫正后的显著阈值为标准,两个性状在全基因组范围内共找到16个显著SNPs及单倍型区域,利用ensembl网站BioMart模块成功鉴定到与这些SNPs或单倍型区域密切相关的10个候选基因。
对于宰前活重,利用SNP和单倍型方法共检测到15个显著SNPs及单倍型区域(表5),分别位于第1、5、6、14、16和17号染色体上,分别邻近或坐落于PPM1K、FAM184B、LCORL、PMP2、R1MS2、PEX14、EPHB3、MPST、RIMS2、SCOC、S100A10、ANGPTL4、FER1L6、FABP4和AARSD1等,其中,3种标记共同鉴定到FAM184B基因,SNP标记和0.3LD单倍型标记共同鉴定到PPM1K。5_SNP_HAP和0.3LD标记共同鉴定到R1MS2基因,0.3LD单倍型标记除了鉴定到SNP标记中的所有候选基因外,还鉴定到PEX14、EPHB3和MPST等基因。本研究表明单倍型方法,尤其是0.3LD单倍型标记鉴定到的基因远远多于SNP方法,且单倍型方法鉴定到的显著性位点或区域大多位于基因内部。
对于屠宰率,共检测到位于第1、14和28号染色体上4个显著SNPs位点(表6),它们邻近或坐落于EPHB3、RIMS2和ANTXRL等基因,其中,SNP和0.3LD标记共同鉴定到EPHB3基因。且所有被鉴定到的显著位点均位于基因的内部。
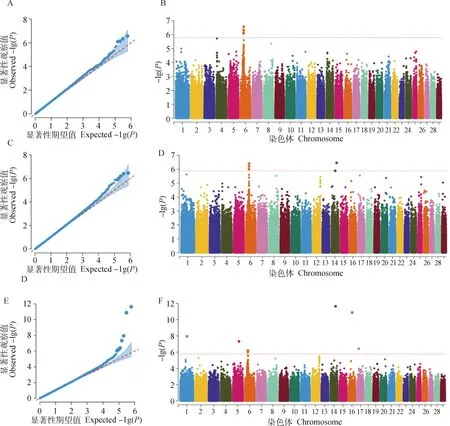
A和B表示利用SNP标记,C和D表示利用5_SNP_HAP单倍型标记,E和F表示利用0.3LD单倍型标记A and B represent SNP marker, C and D represent 5_SNP_HAP haplotype marker, E and F represent 0.3LD haplotype marker 图3 基于SNP、0.3LD单倍型和5_SNP_HAP单倍型标记对宰前活重性状关联分析结果的Manhattan和QQ图Fig.3 Manhattan and QQ plots for the GWAS results of LW by SNP, 0.3LD and 5_SNP_HAP marker
3 讨 论
在肉牛生产中,屠宰性状是备受关注的经济性状,在肉牛产业中发挥着着重要的作用。本研究通过GWAS策略鉴定影响华西牛屠宰性状的分子标记或候选基因,有助于从遗传水平上探索肉牛生长发育的遗传机制,对该品种遗传改良具有重要意义,为后续全基因组选择、分子标记辅助选择等提供可靠的理论基础。本研究基于本团队组建的华西牛资源群体的基因型及表型数据,通过SNP和单倍型标记分别对宰前活重(LW)和屠宰率(DP)等重要经济性状进行了GWAS研究。
通过对群体分层效应进行校正,可有效减少由于遗传背景不同导致的假阳性位点的出现。在做GWAS分析时,选取放入到模型中PCA个数的依据主要有两种,一种是根据经验,选取一定数量的PCA放入模型中[35-38];另外还有根据EIGENSTRAT软件计算各个主成分是否有显著的统计学意义,将P值小于0.05的主成分纳入群体分层校正中[39]。本研究的GWAS结果如图2和图3所示,整体上看,利用单倍型方法进行GWAS鉴定到的基因远远多于SNP方法,且所有SNP方法能够鉴定到的显著性位点,利用单倍型方法也可以鉴定到。在一定程度上说明,利用单倍型进行GWAS可以增加显著性位点的检测能力。这与前人的研究结果一致[14-15]。
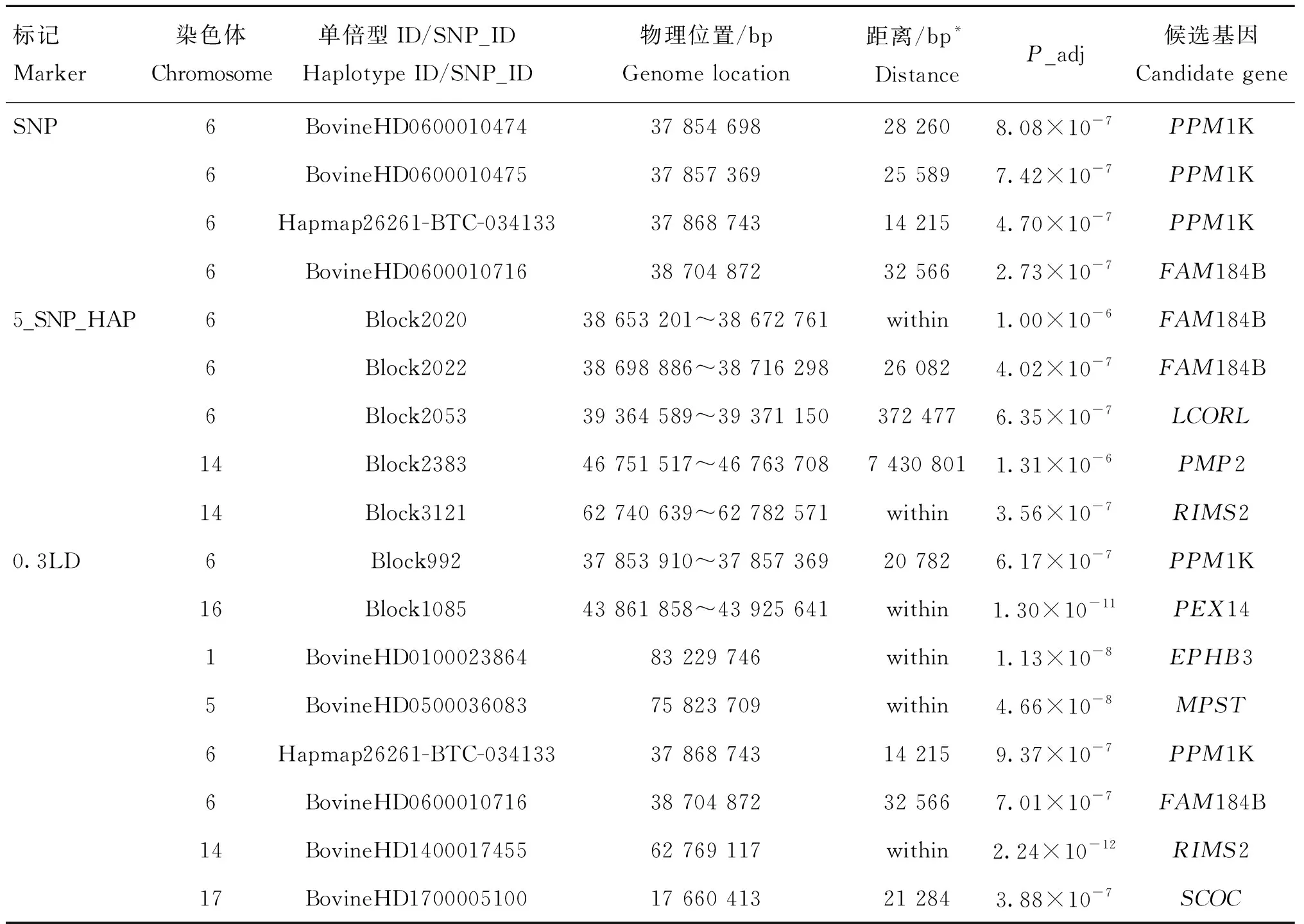
表5 基于SNP、0.3LD单倍型和5_SNP_HAP单倍型标记对宰前活重性状的关联分析结果
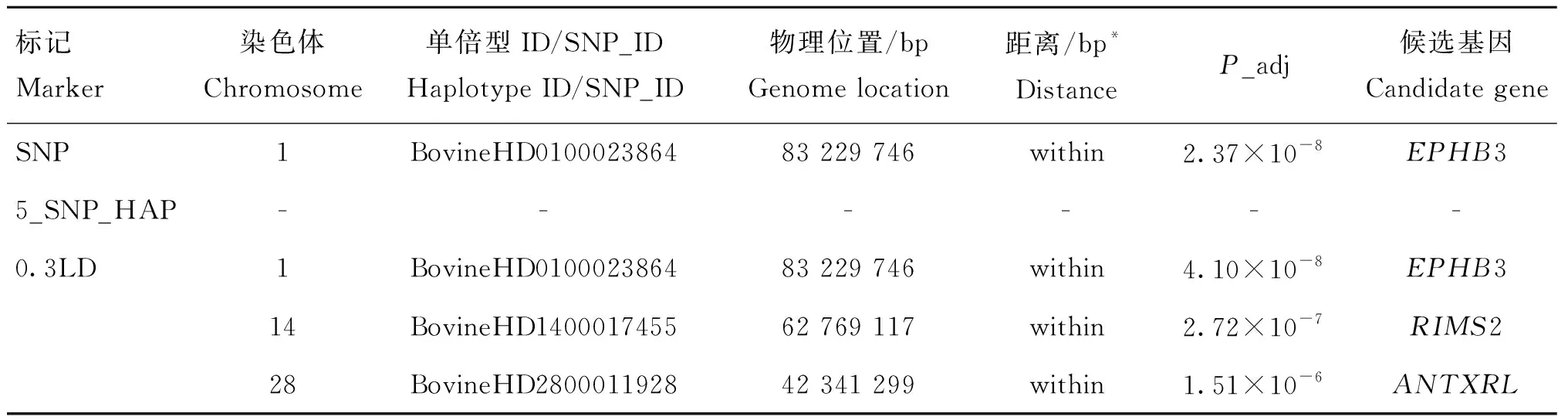
表6 基于SNP、0.3LD单倍型和5_SNP_HAP单倍型标记对屠宰率性状的关联分析结果

A和B表示利用SNP标记,C和D表示利用5_SNP_HAP单倍型标记,E和F表示利用0.3LD单倍型标记A and B represent SNP marker, C and D represent 5_SNP_HAP haplotype marker, E and F represent 0.3LD haplotype marker图4 基于SNP、0.3LD单倍型和5_SNP_HAP单倍型标记对屠宰率性状关联分析结果的Manhattan和QQ图Fig.4 Manhattan and QQ plots for the results of DP by SNP, 0.3LD and 5_SNP_HAP marker
本研究中的3种标记共鉴定到10个与宰前活重和屠宰率等屠宰性状显著相关的候选基因(表5和表6)。其中,SNP标记鉴定到的3个候选基因利用单倍型方法也可鉴定到。许多候选基因已经在其他牛品种或物种上被相继报道。对于宰前活重性状,FAM184B在3种标记中被共同鉴定到。FAM184B是一种蛋白质编码基因,位于6号染色体的38 614 370~38 672 306 bp区域。已有研究表明,FAM184B是一种与生长发育和胴体性状显著相关的候选基因[40]。Xia等[41]在肉牛群体中进行全基因组关联分析的研究中发现,FAM184B是与骨重显著相关的候选基因。表5中的结果显示,PPM1K在SNP标记和0.3LD单倍型标记中被鉴定到,据报道PPM1K是编码Mn2+/Mg2+依赖蛋白磷酸酶的PPM家族成员。Abo-Ismail等[42]在加拿大安格斯、海福特及其杂交群体中进行全基因组关联分析发现,PPM1K是与日增重、代谢体重及皮下脂肪沉积性状显著关联的基因。RIMS2在SNP标记中没有被鉴定到,但在两种单倍型方法同时被鉴定到了。已有报道指出,该基因与胴体重和净肉率性状显著相关[43-46],也有研究认为该基因与骨病显著相关[47]。在5_SNP_HAP单倍型标记中成功鉴定到了LCORL,鉴定到的单倍型区域位于该基因的下游,距离该基因372 477 bp,该基因是与生长发育相关的候选基因,很多的研究报道均已指出LCORL是胴体重、日增重、骨重和眼肌面积等性状重要的候选基因[41,48],尽管该单倍型区域距离LCORL较远,但相比于SNP标记,单倍型方法鉴定到了该基因,在一定程度上证明了单倍型方法具有更强的检测能力。同样在0.3LD单倍型标记中鉴定到的SCOC是与体脂率和体重指数等性状显著关联的候选基因。国内外几乎未见对于屠宰率性状的GWAS 研究,但在本研究中找到EPHB3、RIMS2和ANTXRL等候选基因,据报道EPHB3是重要的蛋白质编码基因,主要在长骨的增殖软骨细胞中持续表达[49]。RIMS2在宰前活重和屠宰率性状中同时被鉴定到,证明其可能是调控生长发育的重要基因。
4 结 论
本研究基于SNP、0.3LD单倍型和5_SNP_HAP单倍型标记分别对宰前活重和屠宰率等屠宰性状进行了GWAS研究。结果发现,3种标记共鉴定到10个与屠宰性状显著相关的候选基因,利用单倍型方法的GWAS鉴定到的基因多于基于SNP标记的GWAS,表明以单倍型开展GWAS可以综合考虑SNP位点间连锁关系,并能较好的揭示复杂性状的遗传结构。
猜你喜欢
军事文摘(2022年16期)2022-08-24
中国现代医生(2022年21期)2022-08-22
今日农业(2021年11期)2021-08-13
智慧健康(2021年17期)2021-07-30
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年4期)2020-12-09
医药前沿(2020年20期)2020-11-10
中西医结合肝病杂志(2020年2期)2020-10-27
三农资讯半月报(2020年2期)2020-03-09
新课程·下旬(2018年9期)2018-11-14
