
基于密度泛函理论的卡博特韦分子反应活性位点预测*
2023-02-21 07:48唐海飞
广州化工 2023年18期
唐海飞,陈 思,王 姣
(湘潭医卫职业技术学院,湖南 湘潭 411104)
艾滋病是由艾滋病病毒(human immunodeficiency virus,HIV)引起的人类严重的传染病之一。2021年,我国艾滋病发病人数为60 154人,死亡19 623人,死亡人数及死亡率在甲、乙类法定报告传染病中都高居榜首[1]。目前,常对感染HIV风险最高的人群应用抗逆转录病毒药物作为暴露前预防。卡博特韦是ViiV Health-care公司生产的第2代整合酶链转移抑制剂。研究表明,卡博特韦(结构见图1)可以长效预防HIV感染[2-3],并于2021年12年20日被美国FDA批准用于高危成人及青少年的暴露前预防[4],预计2022年底在我国获批上市。

图1 卡博特韦分子结构
目前我国学者对该药研究较少,国外对于该药的研究主要集中在临床试验[2-3,5-7]及药理作用[8-9]、药物制剂[10-11]研究等方面。关于卡博特韦药物分子本身反应活性位点的相关研究较少,尚未发现运用量子化学手段对其在分子水平进行结构和反应位点的研究。通过量子化学手段在分子水平研究药物结构及性质,所得结果定量、准确,并可实现可视化,所得结论可为药物深入研究提供重要的参考价值,具有重要意义。
因此,本文以卡博特韦药物分子为研究对象,利用量子化学软件对其分子表面静电势、前线分子轨道、原子电荷、概念密度泛函活性指数进行分析,分析其反应活性位点,为分子水平理解卡博特韦的结构特点、药理作用及构效关系提供理论参考。
1 方 法
依据量子化学密度泛函理论,在B3LYP/6-311++G**方法水平下运用Gaussian 16程序对卡博特韦分子进行结构优化,得到稳定结构及波函数文件,运用Multiwfn 3.8软件[12]对分子表面静电势[13](electrostatic potential,ESP)、前线分子轨道[14]、原子电荷[15]进行了计算分析。同时,基于概念密度泛函理论,也对简缩福井函数等局部描述符和电负性、化学势、化学硬度、化学软度、亲电指数及亲核指数等全局描述符[16]进行了计算分析。
2 结 果
2.1 Mulliken电荷
通过查阅Gaussian 16的分子结构优化文件可以得到除氢原子外的各原子的Mulliken电荷,结果见表1。通过对比表1的数据可知,氮原子及氧原子含有较多的负电荷,碳原子含有较多正电荷,这主要是它们的电负性不同所致。N原子中,吡啶环中N(4)所含负电荷较多;O原子中,羟基O(27)、羰基O(24)含负电荷较多。表明这三处易发生亲电反应。羰基C(14)正电荷在整个分子中最多,因其与O(28)原子相连,受到O(28)原子吸电子诱导效应影响;另外,与羰基C(14)情况类似的羰基C(6)的正电荷也较多。表明这两处易发生亲核反应。这与赵智等[17]分析布洛芬分子波函数时情况相似。

表1 卡博特韦分子中除氢原子外各个原子的Mulliken电荷
2.2 分子表面静电势
卡博特韦分子表面静电势极大值点和极小值点见图2,图2中蓝色为极小值,红色为极大值,静电势极值点编号及数值见表2。
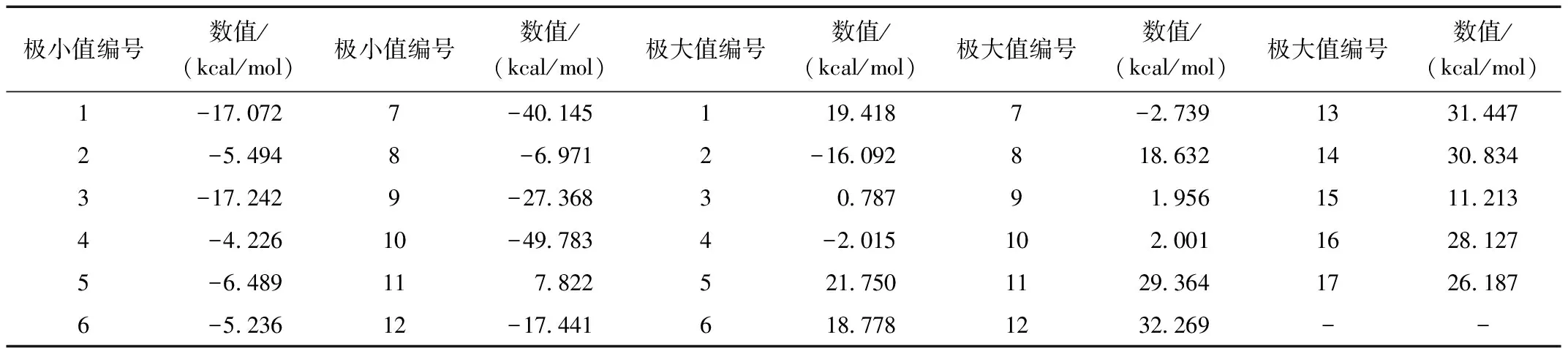
表2 卡博特韦分子表面静电势极值点大小及编号

图2 卡博特韦分子表面静电势极值点分布图
结合图2和表2可知,卡博特韦在分子中O原子及N原子附近主要分布极小值点,极大值点主要分布在H原子附近。这主要是由原子的电负性决定的,电负性越强的原子吸电子能力越强,从而附近呈现极小值点。标号为10的极小值点为整个分子最小的极值点,在羰基O(28)原子及羟基O(27)原子附近;羰基O(23)附近编号7极小值点及羰基O(24)附近编号9极小值点数值也较小。表明这三处电子丰富,在发生静电吸引形成复合物时比较活泼,也可能易发生亲电反应。另一方面,编号为12的极大值点在亚甲基H(31)附近,此处的静电势数值在整个分子中最大。编号为13、14的极大值点静电势数值也较大,分别在嘧啶环H(30)和亚甲基H(32)附近。一般地,复合物的形成由于静电吸引趋向于静电势最大值和最小值相互靠近。
因此,卡博特韦在以氢键、卤键等形式形成复合物时,羰基O(28)、羟基O(27)、羰基O(23)及羰基O(24)及亚甲基H(31)将发挥主要作用。这对研究卡博特韦分子晶体堆积、卡博特韦与受体结合时有重要帮助。
2.3 前线分子轨道
根据前线轨道分子理论[14],最高占据轨道(highest occupied molecular orbital,HOMO)束缚电子能力较差,具有给电子性质,最低未占轨道(lowest unoccupied molecular orbital,LUMO)则性质相反。前线轨道能级差越小,反应活性就越强。卡博特韦的前线轨道结构图见图3,其中蓝色表示波函数负相位,绿色表示波函数正相位。卡博特韦HOMO轨道能量为 -6.678 eV,主要位于羟基O(27)、羰基O(23)、叔胺N(4)附近,表明此处易受亲电试剂攻击,发生亲电反应,与静电势、局部离子化能分析结果一致。LUMO轨道能量为-2.038 eV,主要分布羰基C(6)、吡啶环C(10)、羰基C(9)上,表明这些位置发生亲核反应的可能性稍大。

图3 卡博特韦分子的HOMO和LUMO轨道图
2.4 原子电荷
原子偶极矩校正的Hirshfeld电荷(atomic dipole corrected Hirshfeld atomic charge,ADCH)可表征分子内不同原子所带电荷,带负电荷越多则亲电活性可能越强[15]。表3列出了卡博特韦分子中较大及较小的ADCH电荷值。从表中可以看出,羰基O(23)、O(24)、O(28)和羟基O(27)具有较小原子电荷值,表明这四处具有较好的亲电活性。同时,羰基C(14)、C(10)和羟基H(43)、氨基H(37)具有较大原子电荷值,表明这四个地方易发生亲核反应。这与唐海飞等[18]研究黄曲霉毒素B1分子药物结构和性质时所得结论类似。

表3 卡博特韦部分原子ADCH电荷
2.5 简缩福井函数
简缩福井函数可定量分析分子中原子的亲电亲核反应活性。f+越大,亲核活性越强;f-表示越大,亲电活性越强[12]。由表4可知,卡博特韦分子中,羰基O(24)、羟基O(27)、羰基O(23)、氨基N(4)原子的f-值均较大,表明这四处易发生亲电反应。羰基C(9)、C(6)及吡啶环C(10)原子对应的f+值较大,可能是亲核反应活性位点。
全局活性参数也可用于预测化学反应活性。表5列出了卡博特韦的全局活性参数,在比较卡博特韦与其他药物的反应活性时可以进行参考。卡博特韦亲电指数小,亲核指数大,表明更多的电子将从分子内转移出去,易被亲电试剂进攻,发生亲电反应。该分子硬度大,软度小,表明分子反应活性并不强。

表5 卡博特韦的全局活性参数
3 结 论
卡博特韦分子羟基O(27)原子具有较多的Mulliken负电荷(-0.736 hartree),附近具有分子表面静电势极小点 (-49.783 kcal/mol),具有较高负电荷(-0.317 a.u.),较高f-值(0.059),为HOMO轨道,表明其具有较强的亲电活性,易发生亲电反应。类似的,羰基O(24)及O(23)也为亲电反应活性位点。分子中亲核反应的部位主要集中羰基C(14)、C(6)和C(9)原子上。在静电吸引中羰基O(28)、羟基O(27)及亚甲基H(31)将发挥主要作用。本文全面分析了卡博特韦分子反应活性位点,为进一步理解卡博特韦化学性质、作用机理、构效关系,以及推动卡博特韦的结构改造和进一步开发利用奠定了一定的理论基础。
猜你喜欢
广州化工(2022年19期)2022-11-09
广州化工(2022年18期)2022-10-22
中学生数理化·中考版(2021年10期)2021-11-22
陶瓷学报(2021年5期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
硅酸盐通报(2020年1期)2020-02-25
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
应用化工(2014年1期)2014-08-16
生物加工过程(2013年1期)2013-03-11
