
双钙钛矿卤化物Cs2AgBCl6(B=Cr,Mn,Fe)电子结构和光学性质的第一性原理研究
2023-06-26 09:13郝久源祖宁宁
黑龙江大学自然科学学报 2023年2期
张 敏,郝久源,刘 派,李 瑞,祖宁宁
(齐齐哈尔大学 理学院,齐齐哈尔 161000)
0 引 言
根据现有的报道,Zhou等利用CsCl、AgCl和CrCl3晶体在700 ℃条件下合成了Cs2AgCrCl6,其结构空间群为(R-3m)[8]。研究人员利用第一性原理方法对Cs2AgCrCl6的性质进行了计算,结果表明Cs2AgCrCl6为立方(Fm-3m)结构,且在0~5 eV光子能量内有两个较高的吸收峰[9-11]。Xian等利用CsCl、AgCl和FeCl3晶体通过水热法制备出了Cs2AgFeCl6晶体,实验测量结果表明该晶体为(Fm-3m)立方结构,带隙值为1.55 eV,吸收带边在800 nm左右[12-13]。Dahl等经过对材料Cs2AgInCl6和Cs2AgSbCl6的光吸收实验进行测量,测量结果表明在不同条件下Cs2AgSbCl6均比Cs2AgInCl6吸收光谱所分布的波长长[14]。对比以上报道不难发现,无论是对比Cs2AgCrCl6和Cs2AgFeCl6还是Cs2AgInCl6和Cs2AgSbCl6,材料的吸收光谱均发生了红移现象。
由于双钙钛矿卤化物Cs2AgBCl6(B=Cr,Mn,Fe) 中B位均为磁性离子,参与电子间的相互作用,所以B位离子改变可以影响电子的跃迁几率,同时,改变材料对光的吸收,即B位离子改变是一种可以调控材料性质的手段。综合以上分析,采用第一性原理的研究方法对Cs2AgBCl6(B=Cr,Mn,Fe) 的电子结构和光学性质进行理论计算,并分析B位离子的改变对材料态密度在费米能级附近分布的影响,讨论材料B位离子改变后材料带隙的变化,以及B位离子改变给晶体光吸收带来的变化。
1 计算方法
使用VASP软件包[15-16]对Cs2AgBCl6(B=Cr,Mn,Fe) 的晶体结构进行了优化,使用广义梯度近似(Generalized gradient approximation,GGA)的PBE泛函处理电子间的交换关联能,使用投影缀加平面波方法处理电子与离子间的相互作用[17-18]。为确保自洽计算的准确性,设置平面波截断能为500 eV,总能量收敛精度为10-5eV,相互作用力精度为10-1eV·nm-1。根据Monkhorst-Pack方法对高对称布里渊区的K网格选取为5×5×5。
采用基于全势线性缀加平面波(Full potential linearized augmonted plane wave,FPLAPW)方法的WIEN2K程序计算晶体的电子结构以及光学性能[19-20]。在布里渊区内选取了8 000个K点,设置平面波截断能为7.0,在自洽计算的过程中当能量收敛小于10-5Ry/f.u.时默认为能量收敛。同样使用PBE形式的GGA处理电子的交换关联能。
为了准确模拟电子的真实状态,考虑到d电子库伦关联作用,采取了“旋转不变”法进行“+U”计算(GGA+U)[21]。此外,采用文献[22]给出的线性响应方法计算了体系过渡金属元素的U值,得到Ag的U值在2.00~3.00 eV,Mn的U值在5.00~7.00 eV,Cr的U值在4.00~5.00 eV,Fe的U值在5.00~6.00 eV,这些U值范围与相关研究中所使用的经验值[23-26]是一致的。在计算得到的U值范围内,选取了多组U值对材料性质进行测试,测试结果表明,不同U值对材料的带隙大小、光谱峰值大小和峰值位置有些许影响,但对体系的物理性质所展现出的规律性是没有影响的。因此选取UAg= 2.00 eV,UB= 5.00 eV 的计算结果进行讨论。
2 结果与讨论
2.1 晶体结构
根据文献[9-13]报道,Cs2AgCrCl6和 Cs2AgFeCl6均为立方结构(Fm-3m),基于此,对Cs2AgBCl6(B=Cr,Mn,Fe) 进行了结构优化,得到的结构数据如表1所示,同时表1中也给出了文献报道的Cs2AgCrCl6和Cs2AgFeCl6的结构实验数据。通过对比发现,计算得到晶格常数与实验测量值符合得较好,略大于实验值是因为GGA方法本身会高估晶格常数。三种晶体的晶格常数和Cs—Cl键长几乎相同,但Cs2AgMnCl6晶体的Ag—Cl键长较其他两种材料短约0.01 nm,而Mn—Cl键长却比 Cr—Cl 和Fe—Cl键长长约0.01 nm,产生这种变化的原因将在后面的讨论中分析。

表1 Cs2AgBCl6 (B=Cr,Mn,Fe) 的空间群、晶格常数和键长Table 1 Space groups,optimized lattice parameters and bond distances of Cs2AgBCl6 (B=Cr,Mn,Fe)
另外,对于Cs2AgMnCl6晶体,迄今没有实验报道,所以对其进行了容差因子和八面体因子的计算。容差因子t在1927年由Goldschmidt提出,其公式[27]为:
(1)

μ=rB/rX
(2)
若μ在0.41~0.90,则证明该八面体结构是稳定的。2019年,科罗拉多州立大学Bartel等提出了另一种容差因子τ的表示公式[30]为:
(3)
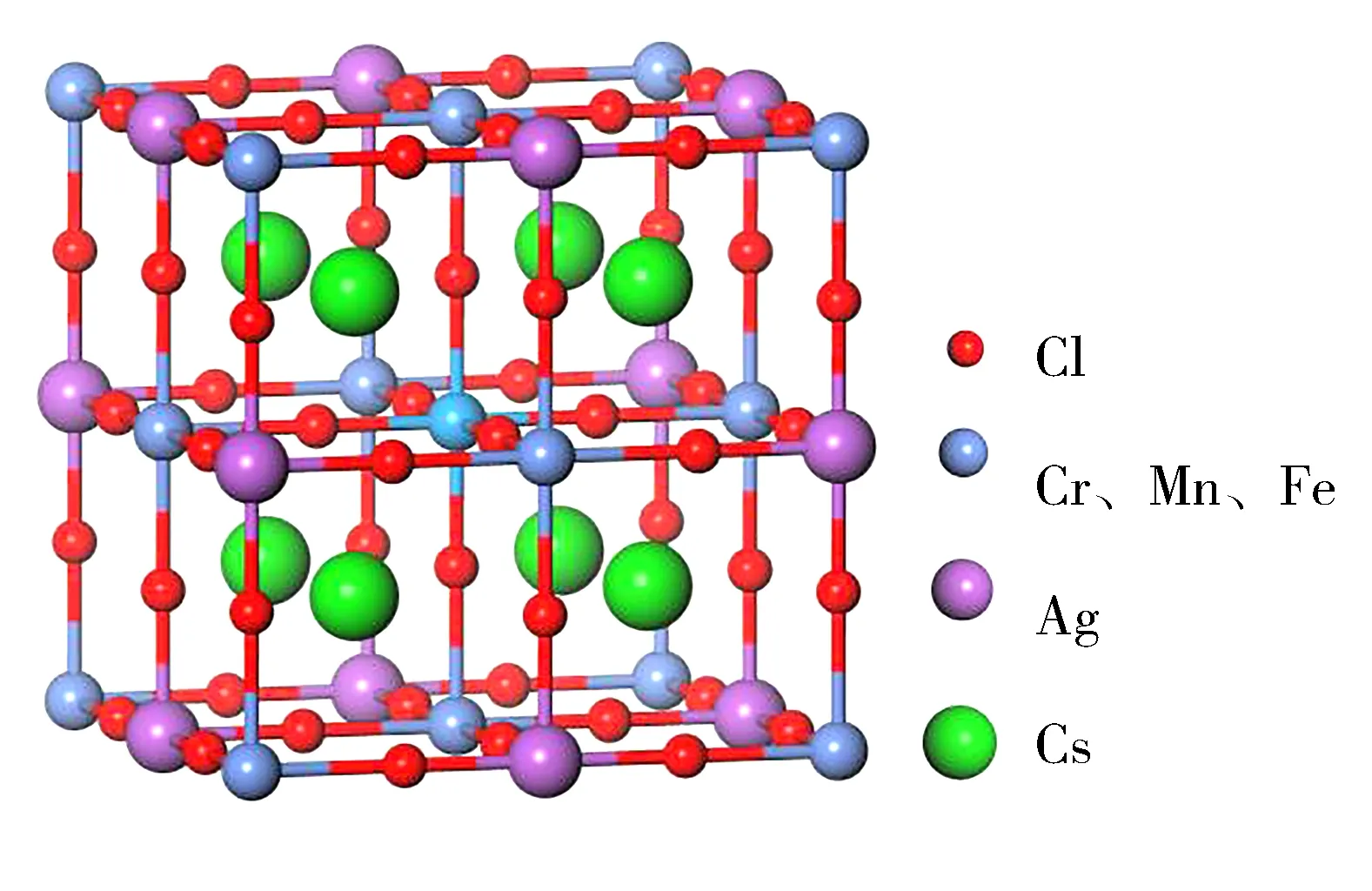
图1 Cs2AgBCl6 (B=Cr,Mn,Fe) 的立方晶体结构Fig.1 Cubic crystal structures of Cs2AgBCl6 (B=Cr,Mn,Fe)
式中rA和rB的物理意义同式(1),nA是A离子的氧化态,对于大多数卤化物和一些氧化物nA=1,在rA>rB并且τ<4.18的材料被预测为稳定的钙钛矿材料。
将Cs2AgMnCl6晶体中各离子半径代入以上公式可得t=0.907,μ=0.498,τ=3.098(Cs、Ag、Mn和Cl离子半径分别为1.670、1.500、0.654和1.810 nm)。通过以上对Cs2AgMnCl6晶体容差因子以及八面体因子的计算说明了理论情况下所建立的Cs2AgMnCl6结构(Fm-3m)的稳定性。另外,利用化合物AgCl、CsCl和MnCl3计算了Cs2AgMnCl6的结合能,为-3.37 eV,这表明Cs2AgMnCl6结构稳定。
2.2 化学键
为了分析Cs2AgBCl6(B=Cr,Mn,Fe) 的电荷分布及元素间化学键的性质,进行了电荷面密度计算,3种材料在(100)面的电荷密度如图2所示。可以看出,Ag与Cl之间以及B位离子与Cl之间的电子密度等高线呈各向异性,表现出明显的共价键特征。此外,根据Ag、Cr、Mn、Fe和Cl的电负性分别为1.93、1.66、1.55、1.83和3.16,Ag与Cl、Cr与Cl、Mn与Cl和Fe与Cl之间的电负性差值均小于1.70,表明Ag—Cl和B—Cl均为共价键[31]。综上所述,Cs2AgBCl6(B=Cr,Mn,Fe)中Ag和B位离子与Cl离子间所形成的键均显示共价键特性。
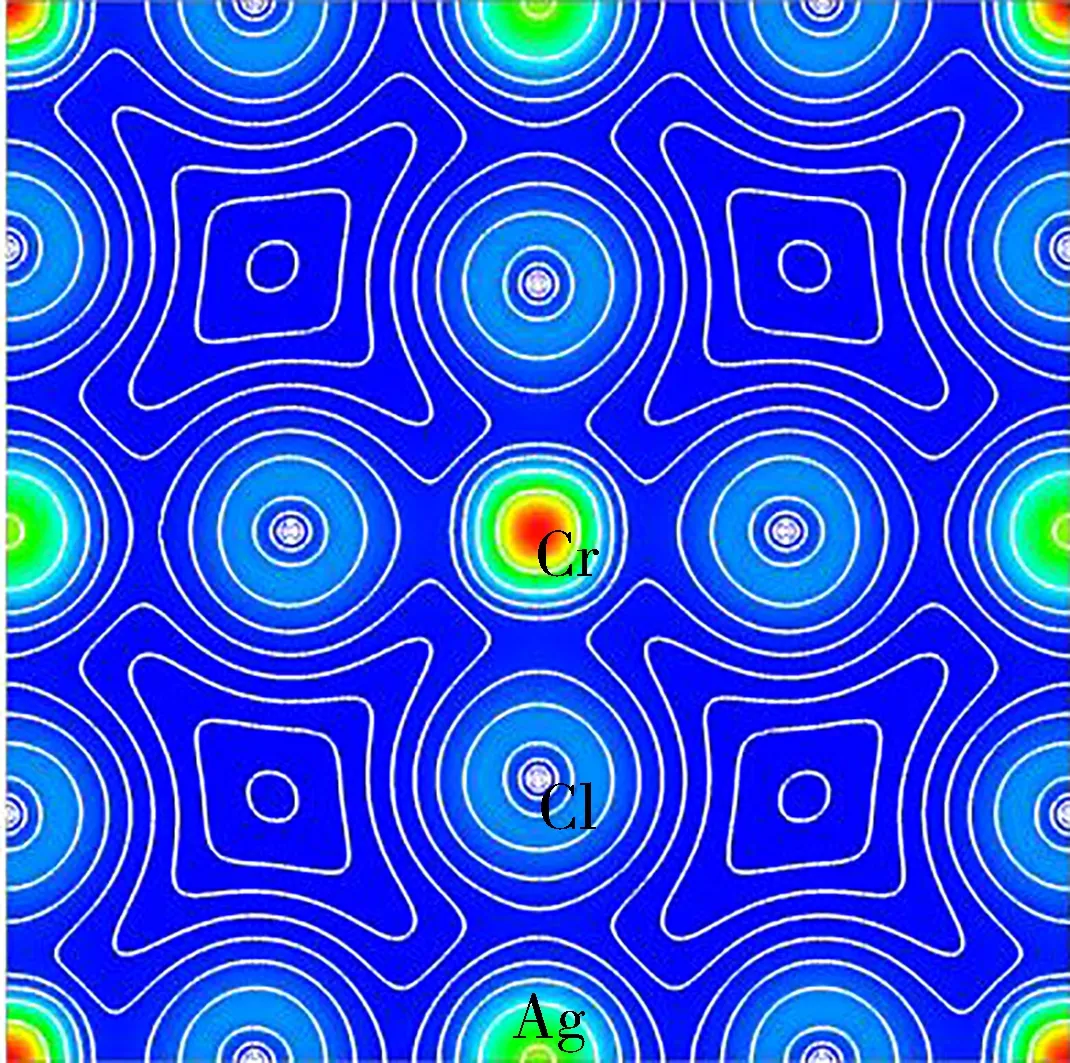
(a) Cs2AgCrCl6
3种材料的差分电荷密度如图3所示,对比图3 (a)、图3(b)和图3(c) 可以看出,Cs2AgMnCl6的Ag与Cl的共价特性强于Cs2AgCrCl6和Cs2AgFeCl6中Ag与Cl的共价特性,反之Cs2AgMnCl6的Mn与Cl的共价特性弱于Cs2AgCrCl6中Cr和Cl以及Cs2AgFeCl6中Fe和Cl的共价性。由于材料的共价特性主要取决于体系中阴阳离子间共用电子对的形成,共价性越强离子间所形成的键越短,这解释了表1中Cs2AgMnCl6的Ag—Cl和B—Cl键长的特性。在Cs2AgBCl6(B=Cr,Mn,Fe) 中,B位离子只与Cl成键。可以看出,B位离子从Cr到Mn再到Fe的改变使得B与Cl之间的共价性先减弱后增强,对比图3 (a) 和 图3(c) 可以看出,Fe和Cl间的共价性略弱于Cr和Cl间的共价性。通过以上分析可以看出,Mn与Cl之间的相互作用最弱,其次为Fe与Cl之间的相互作用,Cr和Cl之间的相互作用最强。

(a) Cs2AgCrCl6
为了验证3种材料中B离子与Cl离子间相互作用的强弱,这里计算了B—Cl键的键布居。键布居的正负分别代表反键态和成键态,绝对值的大小反映了键的强弱,绝对值越大相互作用越强。计算得到的Cr—Cl、Mn—Cl和Fe—Cl键布居分别为-1.03、-0.74和-0.94,表明Mn和Cl、Fe和Cl以及Cr和Cl之间的相互作用依次增强,与差分电荷密度得到的结论相同。
2.3 磁基态
对于Cs2AgBCl6(B=Cr,Mn,Fe) 中的磁性离子B3+,研究了它的自旋排列情况。通过总能计算可知,Cs2AgCrCl6和Cs2AgFeCl6的磁基态均为反铁磁态,即Cr—Cr和Fe—Fe间自旋反向排列,计算得到的自旋磁矩MCr为±3.1μB、MFe为±4.2μB,与文献中报道的计算结果[11,13]是一致的。而Cs2AgMnCl6则为铁磁态,即所有Mn自旋方向相同,磁矩MMn为4.5μB。
2.4 电子结构
计算得到的Cs2AgBCl6(B=Cr,Mn,Fe) 的态密度如图4所示,对于反铁磁体Cs2AgCrCl6和Cs2AgFeCl6,Cr1和Cr2、Fe1和Fe2分别代表晶胞中自旋反平行排列的2个Cr(Fe)离子。由总态密度可以看出,Cs2AgCrCl6和Cs2AgFeCl6为半导体,对于Cs2AgMnCl6,其上自旋方向呈现金属性质,而下自旋方向呈现绝缘性质,因此Cs2AgMnCl6为半金属[32]。对于3种材料的分轨道态密度,可以采用离子模型来分析:Ag离子4d轨道几乎完全被占据,这与其电子组态Ag+(4d10)相对应;Cr3+电子数为3d3,符合图4(a)中Cr离子半个t2g轨道被占据的特点;Mn3+的电子数为3d4,d轨道的占据形式应为t2g3eg1,即eg是半占据的,因此图4(b)中Mn离子的态密度呈现金属性质;Fe3+的电子数为3d5,因此图4(c)中Fe离子的半个d轨道完全被占据。Cs离子的态密度在费米能级附近没有贡献,其主要分布在-6.00 eV附近。由图4可以看出,在-4.00 eV至费米能级的能量范围内,Ag、B的d轨道与Cl的p轨道间存在较强的杂化,而且这种杂化作用在Cs2AgCrCl6中是最强的,这与之前得到的Cr—Cl键最强的结论一致。
布里渊区高对称点为横坐标的Cs2AgBCl6(B=Cr,Mn,Fe) 的能带结构如图5所示,零点处为费米能级。如图5(a)和5(d)所示,对于半导体Cs2AgCrCl6和Cs2AgFeCl6,其上、下自旋方向的电子结构是相同的,所以这里只给出二者其中一个自旋方向的能带结构。Cs2AgCrCl6和Cs2AgFeCl6的带隙宽度分别为1.80和1.34 eV,半金属Cs2AgMnCl6下自旋方向的带隙宽度为3.80 eV,其中Cs2AgFeCl6的带隙宽度与实验测量1.55 eV较吻合[12-13]。由图5可以看出,电子从占据态跃迁到非占据态的难易程度,取决于B在导带中的分布。结合之前从化学键角度对B与Cl之间相互作用的分析,Mn与Cl之间的共价键最弱,即对电子的束缚能力最弱,这与其呈现宏观金属性相对应;其次Cr—Cl共价键强于Fe—Cl共价键,表明电子从占据的Cl_3p轨道向未占据的Cr_3d轨道的跃迁要难于到Fe_3d轨道的跃迁,这与Cs2AgCrCl6的禁带宽度大于Cs2AgFeCl6的禁带宽度吻合。

(a) Cs2AgCrCl6
2.5 光学性质
基于Cs2AgBCl6(B=Cr,Mn,Fe)的电子结构,研究了其光学性质,描述材料光学性质的参数主要包括复介电函数、折射率、反射率和吸收系数。
2.5.1 复介电函数
介电函数是描述材料宏观光学性质的重要参数,复介电函数ε(ω)由实部ε1(ω)和虚部ε2(ω)组成:
ε(ω)=ε1(ω)+iε2(ω)
(4)
式中:实部表示电介质在电场作用下的极化强度,即束缚电荷的能力,数值越大,束缚电荷能力越强,电荷就越不容易极化;虚部表示形成电偶极子消耗的能量,与电子的跃迁有关,电子跃迁是指电子吸收光子能量进而使电子从低能量区的占据态轨道跃迁到高能量区的未占据态轨道。
通过占据态和非占据态波函数的动量矩阵元和Keamers-Kronig关系可以分别得到介电函数的虚部和实部[33]:
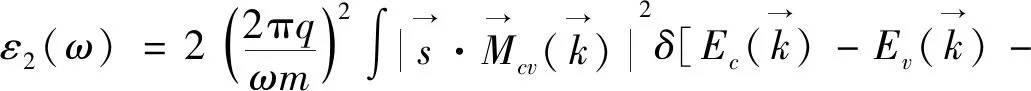
(5)
(6)
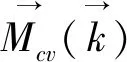
Cs2AgBCl6(B=Cr,Mn,Fe)属于立方晶系,光学性质呈现各向同性,即主介电函数εxx(ω)=εyy(ω)=εzz(ω),本研究仅选取材料的xx方向的光学性质进行分析。Cs2AgBCl6(B=Cr,Mn,Fe)介电函数的实部和虚部随光子能量的变化曲线如图6所示,需要注意的是这里的结果是耦合了上、下自旋得到的总介电函数。由图6(a)可以看出,零频情况下,Cs2AgCrCl6和Cs2AgFeCl6的静介电常数ε1(0)的值分别为3.6和3.8,Cs2AgMnCl6介电函数实部在低能量区域呈现负值,是由于Cs2AgMnCl6为半金属,宏观上为金属性所导致的。根据前面所述,介电函数的虚部ε2(ω)由电子在能带内和能带间的跃迁过程决定。由图6(b)可以看到,半金属Cs2AgMnCl6的ε2(ω)曲线在小于2 eV的低能区具有很高的幅值,就是由于电子在能带内跃迁引起的,除此之外,3种材料的ε2(ω)曲线的特征峰都是电子能带间跃迁的贡献。结合态密度和能带结构,这些跃迁主要来自于占据的Ag_4d、B_3d以及Cl_3p轨道到未被占据的B_3d轨道的跃迁过程。Cs2AgCrCl6和Cs2AgFeCl6的ε2(ω)曲线对光子能量的响应阈值分别约为2.00和1.50 eV,这与前面给出的二者的带隙大小相吻合。

图6 Cs2AgBCl6 (B=Cr,Mn,Fe) 的复介电函数的实部(a) 和虚部(b)
2.5.2 折射率、反射率和吸收系数
晶体的复折射率N(ω)可以表示为:
N(ω)=n(ω)+iκ(ω)
(7)
式中:n(ω)为介质对电磁波的色散,即折射率;κ(ω)为介质对电磁波的吸收的消光系数。它们都可以通过介电函数的实部和虚部计算得到[32]:
(8)
(9)
3种材料的折射率和消光系数随光子能量变化的关系如图7所示。由图7(a)可以得到,Cs2AgCrCl6静态折射率值为1.90,最大折射率在2.20 eV处值为2.52;Cs2AgFeCl6静态折射率值为1.96,最大折射率在2.00 eV处值为2.40;Cs2AgCrCl6和 Cs2AgFeCl6的折射率在所考察能量范围内的变化趋势相同,Cs2AgMnCl6材料的变化趋势不同于其他两种材料的地方是在0 eV附近的变化,这是因为带内跃迁影响着低能量区的电子跃迁情况,所以在0 eV附近Cs2AgMnCl6的趋势不同于其它两种材料是由于Mn_3d轨道上自旋电子的带内跃迁所导致。由图7(b)可以看出,3种材料消光系数随光子能量的变化趋势与介电函数虚部和折射率的变化规律类似,表明他们之间关系相符合。
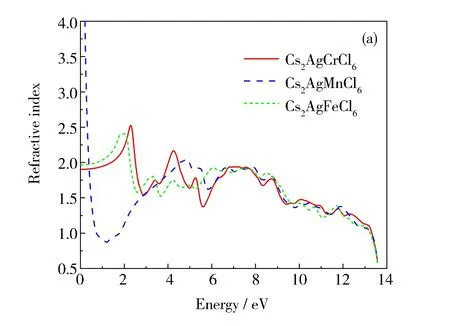
图7 Cs2AgBCl6 (B=Cr,Mn,Fe) 的折射率(a)和消光系数(b)
材料的吸收系数α(ω)可以用以下关系描述[33]:
(10)
式中:c为光速;n为折射率。
由介电函数给出材料的反射率公式R(ω)[32]:
(11)
式中:n为折射率;κ为消光系数。
计算得到的吸收系数在波长300~1 000 nm的变化曲线如图8所示。可以看出,3种材料在这一波段内都有明显的光吸收。Cs2AgCrCl6在400~600 nm波段有一个较宽的吸收峰,在波长493 nm处吸收系数达到峰值23.65×104cm-1。Cs2AgFeCl6则有两个吸收峰,分别在352和533 nm处,峰值吸收系数分别为 20.93×104和21.23×104cm-1,并且Cs2AgFeCl6的吸收带边与之前实验测量的数据相吻合,波长约为800 nm[12-13]。对于Cs2AgMnCl6晶体,相比于前两者它具有更加红移的光吸收,虽然吸收系数偏小,但可以实现在大于600 nm 波长范围内的较宽的光吸收。图8中插图给出了吸收系数随光子能量的变化关系,3条曲线的变化趋势相同,都随着光子能量的提高而增加。Cs2AgCrCl6和Cs2AgFeCl6的吸收阈值分别约为2.00和1.50 eV,这与介电函数虚部ε2(ω)曲线的阈值和带隙宽度是相一致的,而Cs2AgMnCl6在低能区就存在一定的光吸收,这与其金属性吻合。
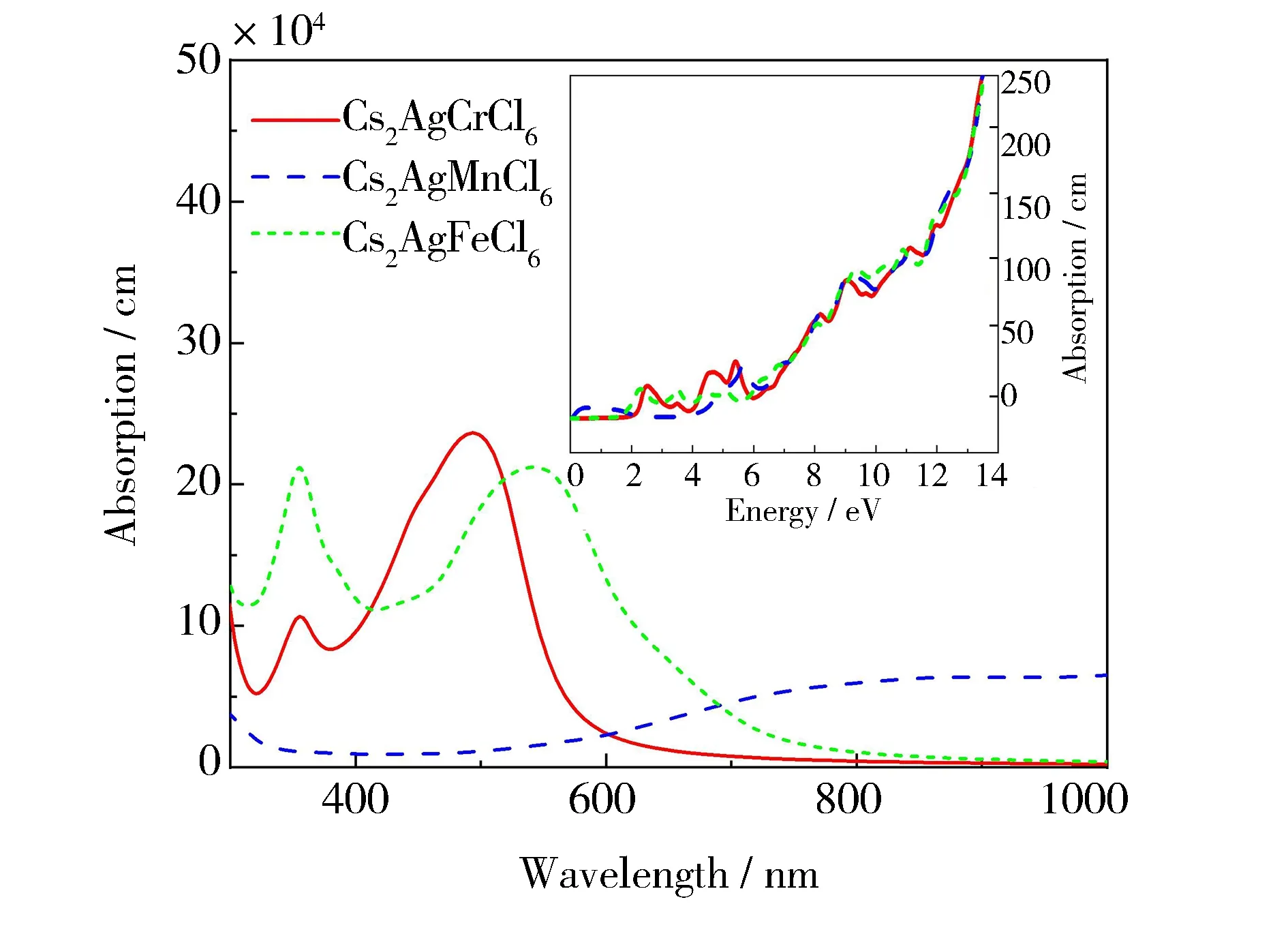
图8 Cs2AgBCl6 (B=Cr,Mn,Fe) 吸收系数随波长的变化(插图为吸收系数随光子能量的变化)Fig.8 Absorption coefficients of Cs2AgBCl6 (B=Cr,Mn,Fe) with the wavelength (the inset illustrates the absorption coefficient with the photon energy)
计算得到的Cs2AgBCl6(B=Cr,Mn,Fe) 的反射率随光子能量变化的关系如图9所示。可以看出,在可见光范围1.60~3.20 eV的3种材料反射率在0.05~0.20。由于在可见光范围内的反射率小于0.25,所以Cs2AgBCl6(B=Cr,Mn,Fe) 无金属光泽,反射损耗较小。

图9 Cs2AgBCl6 (B=Cr,Mn,Fe) 的反射率Fig.9 Reflectivities of Cs2AgBCl6 (B=Cr,Mn,Fe)
3 结 论
采用第一性原理对Cs2AgBCl6(B=Cr,Mn,Fe) 的电子结构和光学性质进行了计算并得出相关结论。总能计算结果表明,Cs2AgCrCl6和Cs2AgFeCl6的磁基态均为反铁磁态,而Cs2AgMnCl6为铁磁态。对Cs2AgBCl6(B=Cr,Mn,Fe) 的化学键分析得出B位原子与Cl原子所形成的键显示共价键特性,其中Mn与Cl之间的相互作用最弱,其次为Fe与Cl之间的相互作用,Cr和Cl之间的相互作用最强。由材料的电子结构可以看出,Cs2AgCrCl6和Cs2AgFeCl6均为半导体,带隙宽度分别为1.80和1.34 eV,Cs2AgMnCl6为半金属。3种材料的带隙宽度的变化规律与B—Cl键间的相互作用强度的变化规律相吻合。复介电函数虚部曲线在所取能量范围内有2个明显的介电特征峰,这些特征峰来自于占据的Ag_4d、B_3d以及Cl_3p轨道到未被占据的B_3d轨道的电子跃迁过程,曲线的阈值光子能量与3种材料的带隙大小相吻合。Cs2AgBCl6(B=Cr,Mn,Fe) 在300~1 000 nm波段都有明显的吸收峰,B位离子按Cr、Mn、Fe的顺序,吸收光谱发生红移,并且3种材料的吸收阈值与介电函数虚部ε2(ω)曲线的阈值一致。反射率计算结果表明,在可见光范围内Cs2AgBCl6(B=Cr,Mn,Fe) 的反射率在0.05~0.20。
猜你喜欢
中学生数理化·高一版(2022年3期)2022-04-15
济南大学学报(自然科学版)(2021年6期)2021-11-10
中等数学(2021年6期)2021-08-14
潍坊学院学报(2021年2期)2021-07-22
高等学校化学学报(2021年7期)2021-07-11
数学学习与研究(2020年23期)2020-01-11
成都信息工程大学学报(2019年3期)2019-09-25
东华大学学报(自然科学版)(2018年1期)2018-06-29
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04

- 黑龙江大学自然科学学报的其它文章
- Crystal structure and terahertz spectrum studies of complex [Cu2(dmp)2(bdppmapy)I2]
- 优化后的QuEChERs方法测定不同基质中百菌清农药残留
- 产胞外多糖酵母菌的筛选鉴定及发酵条件优化
- 低光照环境下基于特征点补偿的图像匹配方法研究
- 一款电池管理芯片的硬件测试平台设计与实现
- The integral equation method for the scattering problem of obliquely incident electromagnetic waves in a chiral medium