
β-MnO2电子结构、磁性、弹性和晶格动力学性质的第一性原理研究
2023-10-26 12:00杨晓翠
白城师范学院学报 2023年5期
杨晓翠
(白城师范学院物理与电子信息学院,吉林 白城 137000)
0 引言
当MnO6八面体单元以不同的方式连接时,MnO2可以形成α-,β-,γ-,δ-MnO2等多种晶型[1].各种类型的二氧化锰及其衍生物具有独特的性能,目前被广泛用于Li/MnO2电池的催化剂、离子筛,特别是作为锂电池的电极材料[2-5].
Mackrodt 等[6]利用第一性原理计算研究了β-MnO2基态的价电荷性质,结果显示Mn 原子的净自旋磁矩约为3 μB,表明未配对的d5电子密度是离域的.Thackeray 等[7]报道了锂离子电池锰氧化物“复合”电极的研究进展,指出设计高锰含量电极是显著降低锂离子电池成本和提高电池性能的一个重要途径.Tompsett等[8]利用带有Hubbard U修正的密度泛函理论(DFT+U)研究了β-MnO2的电化学性能.
众所周知,采用标准局域密度近似(LDA)或广义梯度近似(GGA)处理电子的交换关联势能的密度泛函理论(DFT),在描述具有强局域电子态的磁性材料的物理特性方面存在严重的局限性.这是因为LDA或GGA不能精确地描述局域电子的相互作用[9].最近,科学家们提出了几种方法可以修正采用LDA或GGA计算的DFT的结果,比如,从DFT+U方法[10]到更复杂的自交互修正方法[11].
为了充分利用β-MnO2的各种特性,需要对其电子结构、磁性和晶格动力学性质等有最基本的了解.本文利用带有Hubbard U 修正的密度泛函理论的第一性原理计算,系统地研究β-MnO2晶体的电子结构、磁性、弹性和晶格动力学性质.
1 计算方法
本文利用CASTEP 模拟软件包[12],采用LDA/GGA+U 对嵌在离域态中的局域态进行能量修正.局域态具有较大的库仑作用,可以用U 项来描述处理,而离域态可以用LDA/GGA 来描述[13].LDA/GGA+U 方法的优点是可以在同一计算方案中同时处理离域导带电子和局域d/f电子.实验证明,LDA/GGA+U 方法对于库仑相互作用强的局域电子体系的电子结构计算是非常有效和可靠的[14].
几何结构优化选择GGA+U(Mn原子采用U=2.5 eV),超软赝势,收敛标准采用动能截断值为380 eV,k点网格为3×3×5.弹性常数的计算选用LDA+U 和模守恒赝势的电子自旋极化,收敛标准采用动能截断值为830 eV,k点网格为3×3×5.晶格动力学性质计算选择GGA+U 和不带电子自旋极化的模守恒赝势,收敛标准采用动能截断值为830 eV,k点网格为3×3×5.计算中,自洽收敛精度设置为5.0×10-6eV/atom,原子间力收敛判据为0.1 eV/nm,最大位移为5.0×10-5nm,应力设置为0.02 GPa.
2 结果与讨论
2.1 磁学性质
由于磁序、能量和结构之间的耦合,首先要确定磁序结构的模型.由于Mn4+是一种磁性离子,因此在锰氧化物电子结构研究中计及磁性效应至关重要.以前的研究表明,交换关联方法对锰氧化物有很大的影响[15-16].
锰氧化物的电子态中最显著的特征之一是两种类型的d电子共存,即局域电子和巡游电子.局域电子的磁矩,通过Hund规则与巡游电子强耦合[17].这种双交换相互作用产生铁磁性.
用GGA+U 方法计算了几种磁序模型的单元胞能量,找到了能量最低的磁态.结果表明,β-MnO2是一种亚铁磁性半导体,每个晶胞磁矩为6.0 μB.Mn原子的净自旋磁矩为3.0 μB,与Mn4+价态相关,表明未配对的d5电子是离域的.
2.2 β-MnO2晶胞参数和电子性质
表1 给出了β-MnO2晶胞参数的计算结果以及实验结果,以便于进行对比.GGA+U 预测的晶胞参数与已有的实验和理论结果基本一致.由表1可以看出,采用GGA+U计算结果往往略大于实验测量值.
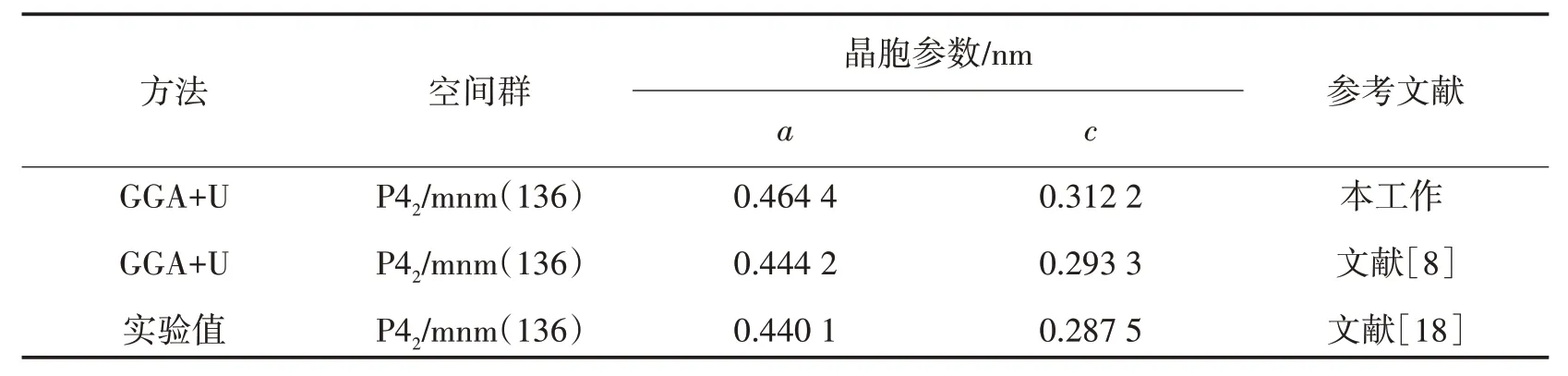
表1 β-MnO2晶胞参数的计算结果以及实验结果
图1 给出了计及自旋极化的β-MnO2能带结构计算结果.从图1 可以看出,在费米能级附近不同自旋方向的能带结构存在较大的分裂.与电子自旋向下态相比,自旋向上态所有的能态(包括导带和价带区域)都向上移动.在费米能级附近,自旋向上态在Г点存在0.7 eV 的直接能隙.考虑到两种自旋状态,研究结果表明,β-MnO2是直接带隙为0.3 eV的半导体,与文献[16]的研究结果一致.
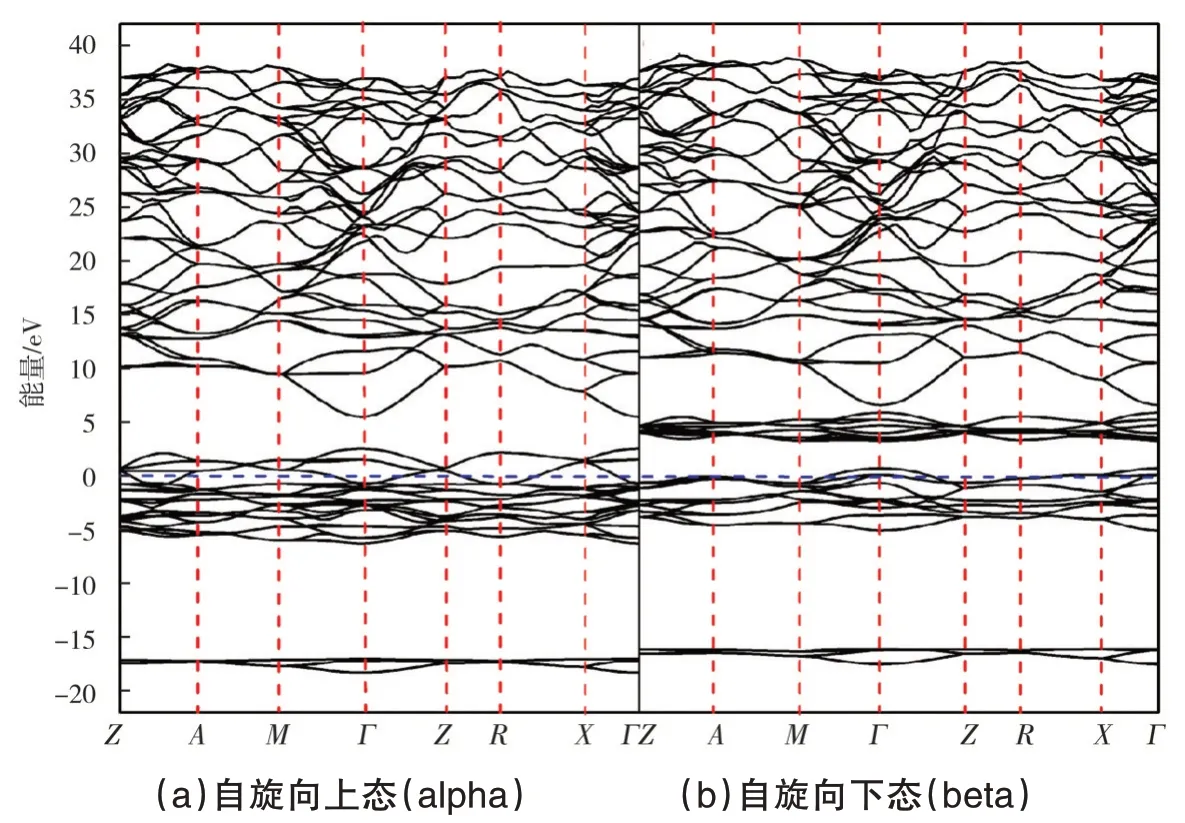
图1 β-MnO2布里渊区的高对称点的自旋极化能带结构
由图1(a)和图1(b)可以看出,能量在5.0 eV 以上的大部分能带结构拓扑是相同的,这反映了成键态的共同起源[19];自旋向下态在4.0 eV 附近的能带结构的平坦性表明Mn 原子的局域d电子起主导作用.自旋向上态和自旋向下态的能带结构主要区别在于Mn 原子的d电子产生的能带部分,这表明存在较大的Mn的d电子交换分裂.
为了理解自旋极化的行为和深入了解磁性的起源,本文研究了β-MnO2的电子结构和偏态密度,计算结果分别如图2和图3所示.

图2 β-MnO2晶胞中O原子的2s和2p电子的偏态密度
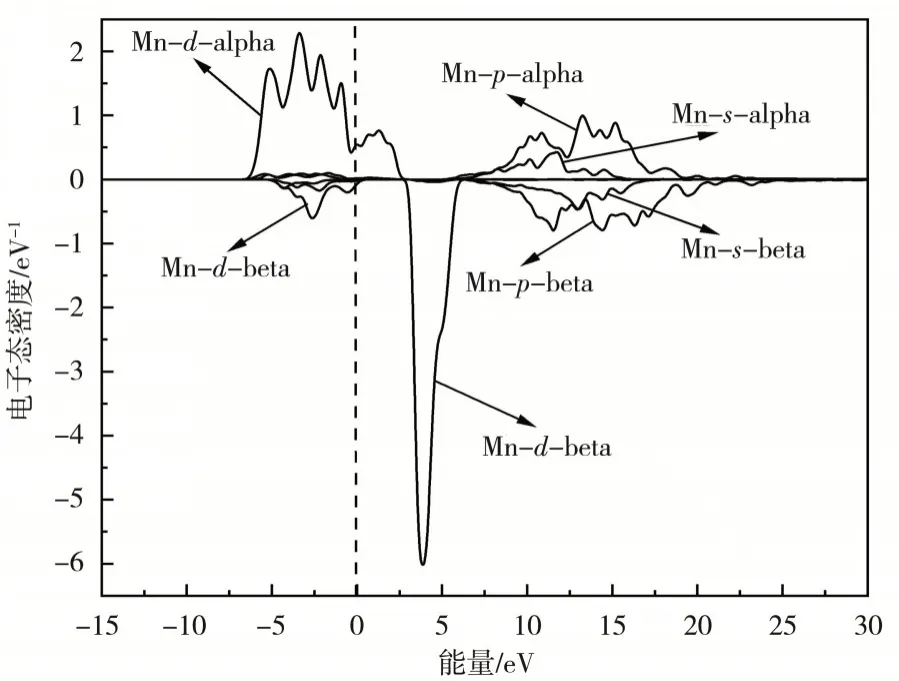
图3 β-MnO2晶胞中Mn原子的3p,3d和4s电子的偏态密度
从图2可以看出,在-18 eV附近两个自旋状态都有较小的交换分裂的O原子的2s电子能带;在费米面之下,这些态主要是由两个自旋状态中没有分裂的O 原子的2p电子贡献的.这表明O 原子的s,p电子对磁性的贡献几乎为零.
从图3 可以看出,Mn 原子的3d电子自旋向上态大部分分布在费米能级以下,而自旋向下态大部分分布在费米能级以上.这种分布说明Mn原子的3d电子是β-MnO2晶胞磁性的主要起源.
2.3 β-MnO2晶体的弹性性质
弹性常数Cij(i,j=1,2,3,4,5,6)描述晶体对外界应力的响应,在许多与材料力学性能相关的实际应用中Cij是必不可少的力学参数.为了验证β-MnO2的力学结构稳定性,采用应变-应力法计算其弹性常数.对优化后的原子施加小的有限应力,优化原子的位置,根据结构的应变求出弹性常数[20].表2列出了环境压力下β-MnO2晶体弹性常数的计算结果.

表2 环境压力下β-MnO2晶体弹性常数Cij(GPa)的计算结果
对于四方晶系,其力学结构稳定性判据为
由表2可知,C44<0,可见,弹性常数不满足力学结构稳定条件,表明β-MnO2晶胞结构在环境压力条件下不是力学稳定相.
2.4 β-MnO2晶体的晶格动力学性质
固体的许多物理性质取决于它们的声子性质,如比热、热膨胀、热传导等.本文计算了β-MnO2晶体的声子谱,环境压力下β-MnO2晶体的沿高对称方向的声子色散曲线和声子态密度如图4所示.

图4 环境压力下β-MnO2晶体的声子色散曲线和声子态密度
从图4 可以看出,虚频率出现在Z,A,R和M高对称方向,这表明β-MnO2晶胞结构在环境压力下不是动力学稳定结构,C44<0是声子谱中出现虚频的原因.
3 结论
本文采用LDA/GGA+U 方法对β-MnO2的基本物理性质进行了系统的研究.电子结构和磁性能结果表明,β-MnO2是直接带隙为0.3 eV 的半导体,每个晶胞磁矩为6.0 μB.Mn 原子的3d电子的自旋向上态和自旋向下态之间存在较大的能带分裂,这是导致晶胞磁性存在的主要原因.弹性常数的计算结果表明,β-MnO2结构在环境压力下不是力学稳定相.声子色散曲线结果表明,β-MnO2的结构在环境压力下不是动力学稳定结构.
猜你喜欢
高中数理化(2022年16期)2022-09-14
汽车实用技术(2022年5期)2022-04-02
高中数理化(2022年4期)2022-03-14
今日农业(2021年7期)2021-07-28
舰船科学技术(2021年12期)2021-03-29
北京汽车(2021年1期)2021-03-04
风流一代·青春(2020年6期)2020-06-19
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
山东化工(2019年1期)2019-01-24
三峡大学学报(自然科学版)(2017年1期)2017-03-20
