
虚拟筛选结合活性验证发现新型SHP2催化位点抑制剂
2023-10-30 13:39黎娅琳任吉霞
聊城大学学报(自然科学版) 2023年5期
黎娅琳,任吉霞
(聊城大学 生命科学学院,山东 聊城 252059)
1 引言
SHP2(SH2 domain-containing protein-tyrosinephosphatase-2)由PTPN11基因编码,属于非受体蛋白酪氨酸磷酸酶。完整的SHP2蛋白晶体结构显示[1],在本底状态下,SHP2通过N-SH2域和催化位点PTP域间形成分子内相互作用而处于一种自抑制的失活构象;在生长因子、细胞因子等外界环境因子的刺激下,SHP2的失活状态被打破,变成有活性的开放构象。研究表明,SHP2参与多种信号通路,SHP2的活化是Ras/ERK1/2通路完全激活所必需的,此外,SHP2活化PI3K/AKT,JAK/STAT,JNK和NF-κB信号通路,进而调控细胞增殖、分化、细胞周期维持和迁移等生理功能[2]。SHP2还通过其N-SH2结构域结合免疫检查点蛋白PD-1的磷酸酪氨酸基序(pTyr motif),参与调节 T 细胞的活性[3]。综上SHP2是细胞中多种信号通路的重要信号调节分子,同时SHP2还是控制细胞因子产生及免疫细胞反应的重要调控因子。
所以,当PTPN11基因发生突变而表达异常时,将导致细胞增殖分化异常,引起多种疾病的发生发展。近几年大量遗传学和临床研究表明,在多种恶性血液病和实体瘤中都存在SHP2突变活化或过表达[4]。例如,生殖系获得功能性PTPN11基因突变会导致Noonan 综合征(40%~50%),该疾病是一种可增加恶性肿瘤风险的常染色体显性遗传发育障碍症[5]。SHP2高活化突变出现于幼年慢性骨髓单核细胞性白血病(JMML,35%),急性B淋巴细胞白血病(7%)和急性髓性白血病(4%)[6,7]。白血病相关PTPN11E76K突变会促进骨髓增殖性肿瘤的发生和发展[8]。SHP2过表达在多种实体瘤中非常常见,例如:在大约55%原发性乳腺癌中SHP2过表达,并会导致预后不良[9]。最近Hana Algül课题组发现KRAS突变驱动的胰腺导管腺癌和非小细胞肺癌依赖于SHP2[10]。据Rene Bernards课题组报道SHP2是KRAS突变型非小细胞肺癌所必需的[11]。除此之外,近期研究表明 SHP2与肿瘤免疫调节密切相关,例如,Zhao等研究发现[12],抑制SHP2活性可激活抗肿瘤免疫力,并可以与阻断PD-1起协同作用,这为肿瘤免疫治疗提供了新的思路。以上研究均提示SHP2是一个抗肿瘤新靶标。靶向抑制SHP2可双管齐下,既减缓癌细胞生长,同时也调节免疫功能以激活其抗肿瘤作用。
综上,研发SHP2小分子抑制剂,对靶向SHP2的抗肿瘤治疗及其分子机制研究都具有十分重要的意义。目前已经有一些 SHP2小分子抑制剂被报道,这些小分子抑制剂主要分为两大类:催化位点抑制剂和变构位点抑制剂[13,14]。张仲寅课题组开发了多个SHP2催化位点抑制剂[15-17],最近,国内学者王文龙课题组[18]和王润玲课题组[19,20]也发现多个新型SHP2催化位点抑制剂。除此,还有4款小分子变构抑制剂进入临床研究[21,22]。
计算机辅助药物设计(computer aided drug design,CADD)技术已在药物研发中得到广泛应用,据估计 CADD 使得新药研发周期缩短了0.9 年,研发费用降低 1.3亿美元[23]。根据药物作用靶点三维结构是否已知,CADD 大概可以分为基于配体的药物设计(如药效团模型)和基于结构(如分子对接)的药物设计。研究证明三维药效团模型和分子对接技术的联合应用可以缩短大型化合物数据库虚拟筛选的时间,并可提高命中率[24]。虚拟筛选获得化合物的活性基本上在 μmol/L 水平,有少量的化合物达到 nmol/L 水平,这一活性水平的化合物可以作为先导化合物进行结构改造,以获得活性高的类药性化合物[25]。计算机辅助药物分子设计作为创新药物研究的新方法和新技术,已经成为一种实用化工具,加入到了创新药物研究的工作流程中,成为现代药物研发的核心技术之一。
本研究使用DS软件构建SHP2催化位点抑制剂和非抑制剂的贝叶斯分类模型,抑制剂的3D-QSAR药效团模型,利用贝叶斯分类模型对大型商业数据库进行筛选,预测阳性化合物再由药效团进行筛选,通过两轮筛选的化合物利用分子对接技术对接到SHP2催化活性位点,通过分析对接结果选出潜在活性化合物,再从公司购买化合物实体,进行酶水平抑制活性检测,最终得到3个具有一定潜在选择性的SHP2新型抑制剂。并通过分析活性最高化合物和药效团匹配和分子对接结果,阐明化合物起抑制作用的机制。
2 材料和方法
2.1 搜集SHP2催化位点小分子抑制剂
从BindingDB数据库下载已报导的SHP2小分子抑制剂的SDF文件[26],利用Discovery Studio 3.1(DS 3.1)打开文件,根据文件中提供的每个化合物的信息,删除非活性位点抑制剂。利用DS 3.1的“Prepare Ligands”模块对剩余SHP2催化位点小分子抑制剂进行去重处理;将去重处理后的所有SHP2小分子抑制剂按照活性IC50进行排序,规定IC50< 20 μmol/L为SHP2的抑制剂,添加标注为1,IC50> 20 μmol/L为非抑制剂,添加标注为-1。
2.2 构建SHP2催化位点小分子抑制和非抑制剂二分类贝叶斯模型
利用DS 3.1的“Generate Training and Test Data”模块,构建训练集和测试集,将注有标记的SHP2催化活性位点抑制剂作为输入化合物,“Split Method”参数设置为“Random”,“Training Set Percentage”参数设置为80。利用DS 3.1中“Create Bayesian Model”模块,以生成的训练集和测试集化合物作为输入,构建贝叶斯模型,设置“Calculate Properties”时,先确定所用分子指纹的类型,然后将确定的分子指纹和常见的几种分子描述符组合进行模型构建。为确定关键分子描述符,采取逐一排查法,最终确定分子指纹和分子描述符的最优组合。
2.3 构建SHP2催化位点小分子抑制剂3D-QSAR药效团
2.3.1 训练集化合物的选取。药效团建模过程中,训练集分子的挑选对模型的构建具有非常重要的作用。利用DS3.1构建3D-QSAR药效团模型对训练集分子有如下要求[27]:(1)18-25个结构多样性的化合物,为保证结果具有统计学意义,化合物分子数目不得低于15个。(2)化合物活性跨度至少为4个数量级,每个数量级至少有3个化合物。(3)结构相似的化合物活性至少相差一个数量级,有相似活性的化合物结构必须不同。(4)所有化合物提供的信息不得冗余。根据这4条要求,从搜集的SHP2催化位点抑制剂中选择16个化合物构成训练集。
2.3.2 3D-QSAR药效团的构建。利用DS 3.1中的“3D QSAR Pharmacophore Generation”模块以16个训练集化合物作为输入化合物,两个高活性化合物1(IC50:0.2 μmol/L)和2(IC50:0.45 μmol/L)用作建模过程中的模板分子,其对应的参数“Principal”及“MaxOmitFeat”分别设为2和0,其余化合物的这两个参数都设为1。
所有SHP2催化活性位点抑制剂利用DS 3.1的“Prepare Ligands”模块进行能量优化,使其初始构象处于能量较低的状态。在“3D QSAR Pharmacophore Generation”模块中“Conformation Generation”参数设为Best,并将能量阈值设为“20 kcal/mol”,然后利用Poling算法对每个化合物产生255个构象,这些构象涵盖了所有可能与SHP2催化结构域结合的化合物的活性构象。“Maximum Pharmacophores”参数设置为5,“Minimum Interfeature Distance”设为1.5,利用剩余SHP2抑制剂作为测试集化合物,“Fitting Method”设置为flexible,“Fisher Validation”设置为95%。除此,构建药效团所需其它参数采用默认参数。
通过对所有SHP2小分子抑制剂的观察和相关文献报道,选择以下药效特征元素作为初始药效特征:氢键受体(Hydrogen-bond acceptor,A),氢键供体(Hydrogen-bond donor,D),疏水特征(Hydrophobic feature,H)),芳香环特征(Ring aromatic feature,R)和电离后呈负电荷基团(NEG_ionizable,N)。
2.3.3 3D-QSAR药效团的评价。通常通过分析计算结果中给出的cost值和其它相关统计学参数对3D-QSAR药效团模型进行初评,对通过初评的药效团模型再利用测试集和交叉验证法进行进一步的评价。参数修改后运算给出最好的5个药效团模型及其相应参数,这些参数主要有:Fixed cost,理想模型的cost值;Null cost,无意义模型的cost值,即模型预测活性和实验活性完全不相关时的cost值;Total cost,对应于每个不同的药效团模型其值不同;相关系数(correlation coefficient),代表利用药效团模型预测的训练集分子活性值和实际实验活性值的相关度;Config值,代表药效团模型空间构象的复杂程度;还有均方根偏差值(RMSD value)等。一个好的药效团模型,应当具有较高的相关系数,其Total cost值应该接近Fixed cost值远离Null cost值,Config值不大于17,均方根偏差越小越好。交叉验证法可以进一步评价通过初评药效团模型的可信度(confidence level),将置信度设为95%,那么程序将产生19个随机表单,每个表单中化合物的结构和活性数据是随机混乱匹配的,但是表单中的其它参数与构建初始药效团模型的参数值完全相同。通过分析随机产生的19个训练集构建的95个药效团模型的统计学参数可评价构建的定量药效团模型的可信性。在测试集验证方法中,将除训练集之外的66个化合物组成独立测试集,运用构建的定量药效团模型对测试集化合物进行活性预测并与其实验活性做回归分析,以验证药效团模型的预测能力。
2.4 分子对接研究
本研究中的所有对接研究均使用GOLD 5.1来完成[28]。SHP2催化结构域的晶体结构来源于蛋白质数据库(PDB ID: 4RDD),因为其在所有的SHP2催化结构域晶体结构中分辨率最高(0.16 nm),所以被作为对接研究中小分子化合物的受体。通过使用DS 3.1将氢原子添加到蛋白质,然后分配ChARMM力场。结合位点被定义为一个球体,其覆盖以配体为中心1 nm范围内的受体氨基酸残基,这个球体足够覆盖活性位点中配体化合物的结合区域。通过将与SHP2催化结构域共结晶的抑制剂对接到受体的活性部位,预先优化了评分函数和对接参数。
2.5 新型SHP2催化位点抑制剂的虚拟筛选
使用构建好的贝叶斯分类模型通过DS 3.1中“Calculate Molecular Properties”模块对大型商业化合物数据库里的化合物进行预测,利用预测为阳性的化合物,构建药效团筛选数据库。使用DS 3.1中的“Search 3D Database”模块,利用建立的药效团模型对构建好的化合物数据库进行3D结构匹配,选择匹配值(Fit Value)超过一定阈值的化合物进行分子对接研究,通过对对接结果的分析,选择购买数个化合物进行后续的活性研究。
2.6 酶水平化合物活性检测
本研究中酶水平活性检测选用欧洲Eurofins Discovery公司的Discovery Services模块中的PhosphataseProfilerTM完成,该方法使用荧光法测试磷酸酶活性。参考文献[29,30]并作适当调整,以6,8-二氟-4-甲基-7-羟基香豆素磷酸酯(DiFMUP)作为底物,SHP2催化其水解产生6,8-二氟-4-甲基-7-羟基香豆素(DiFMU),通过Thermo Scientifific Verioskan Flash多功能读数仪,以358 nm为激发波长来检测450 nm处的荧光值,确定SHP2的酶活。以矾酸钠(Na3VO4·12H2O)为阳性抑制剂,它能够氧化蛋白酪氨酸磷酸酶的保守催化活性位点半胱氨酸,是一种PTPs的通用抑制剂。除了感兴趣的待测化合物被正钒酸钠取代,阳性对照孔包含了反应的所有组分以及控制溶剂效应的DMSO;除了化合物,空白孔包含了反应的所有成分。各种不同酶的活性抑制实验具体操作如下。
2.6.1 SHP2活性检测。使用在大肠杆菌中表达的人重组磷酸酶PTPN11(SHP-2)。将试验化合物与45 ng/mL酶在37 ℃、pH 7.2的改良HEPES缓冲液中预孵育15 min。通过添加100 μmol/L DiFMUP 60 min来引发反应。在358 nm/450 nm下用荧光光谱法读取所形成的DiFMU的量的测定。化合物在10 μmol/L下进行筛选。
2.6.2 SHP1活性检测。使用在大肠杆菌中表达的人重组蛋白磷酸酶SHP1。将试验化合物与2 U/mL酶在37 ℃、pH 7.2的改良HEPES缓冲液中预孵育15 min。通过加入100 μmol/L DiFMUP 30 min来引发反应。在358 nm/450 nm下用荧光光谱法读取所形成的DiFMU的量的测定。化合物在10 μmol/L下进行筛选。
2.6.3 PTP1B活性检测。使用在大肠杆菌中表达的人重组蛋白磷酸酶PTP1B。将受试化合物与170 μU/mL酶在37 ℃、pH 7.2的改良HEPES缓冲液中预孵育15 min。通过加入10 μM DiFMUP 60 min来引发反应。在358 nm/450 nm下用荧光光谱法读取所形成的DiFMU的量的测定。化合物在10 μmol/L下进行筛选。
2.6.4 DUSP22检测。使用在大肠杆菌细胞中表达的人重组蛋白磷酸酶DUSP22。将受试化合物与2 μg/mL酶在37 ℃、pH 7.2的改良HEPES缓冲液中预孵育15 min。通过加入10 μmol/L DiFMUP 30 min来引发反应。在358 nm/450 nm下用荧光光谱法读取所形成的DiFMU的量的测定。化合物在10 μmol/L下进行筛选。
3 结果和讨论
3.1 SHP2催化位点小分子抑制和非抑制剂二分类贝叶斯模型
总共搜集到376个SHP2催化位点抑制剂,其中220个化合物活性(IC50)高于20 μmol/L,在模型构建中作为非活性化合物,标注为-1;剩余157个化合物活性(IC50)低于20 μmol/L,在构建模型时作为活性抑制剂,标注为1。训练集化合物占比80%,剩余20%为测试集化合物,经程序随机分配后,训练集总共301个化合物,其中178个非活性化合物,123个活性化合物;测试集总共75个化合物,由42个非活性化合物和33个活性化合物组成。
模型构建时,分子指纹LCFC16与分子描述符Num_H_Donors,Molecular_Fractional Polar Surface Area为最优组合,给出最好的二分类结果。最优贝叶斯分类模型SHP2_BDM评价数据见表1,ROC Score,SE,SP和Concordance数值越接近1,说明模型的二分类能力越优。从表1中的数据可见已构建模型是一个优秀的贝叶斯分类模型,可用于后续虚拟筛选。

表1 SHP2催化位点抑制剂和非抑制剂贝叶斯分类模型的验证结果
3.2 3D-QSAR药效团模型的构建及评价
3.2.1 药效团模型的构建。利用选出的16个训练集小分子化合物(图1),经过运算得到5个最佳的药效团模型,其参数见表2,从表2数据可见,P1-P5的Cost差值分布在53.014~63.656之间。就药效团有意义的显著性而言,真正重要的是任何药效团的Total cost与Null cost之间的差异大小,如果二者差异为40~60,那么代表有75%~90%的可能性活性数据和化学结构是真正相关的。Total cost和Null cost差值越大代表构建的药效团预测化合物活性的能力越高。

图1 构建药效团模型的训练集分子化学结构及其生物活性(IC50 : μmol/L)
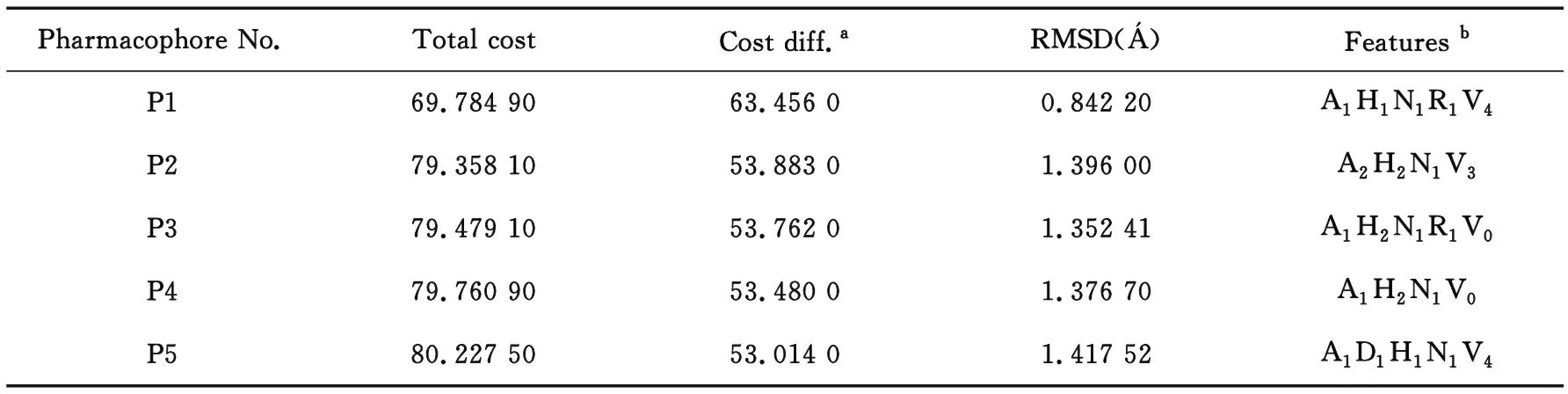
表2 排名前五的SHP2催化位点小分子抑制剂定量药效团模型的统计学参数
图2(a~e)显示了构建的5个定量药效团模型,包括药效特征元素种类和药效团特征元素三维空间以及距离限制,图2(f)显示了5个药效团的叠合结果,从图2可以看出5个药效团的药效特征的空间排布和各个特征元素之间的距离是相差无几的。但5个药效团的药效特征的种类和数目却有较大差别。5个药效团都含有空间分布类似的1个电离负电荷基团特征,1个氢键受体和1个疏水特征。P2、P3和P4还各含有一个位置相似的疏水基团特征;P1和P3还各含有一个位置类似的芳香环特征;P2在与P1和P3芳香环特征位置相似的空间位置有一个氢键受体;P5还在一个靠近电离负电荷特征的位置有个氢键供体;除此,P1、P2和P5还含有3~4个不等的排斥球,即化合物不能占据的空间。经过分析5个药效团含有的药效特征种类、数目和空间排布,本研究倾向选择P3和P5进行后续的活性化合物筛选。
3.2.2 药效团模型的验证。好的药效团模型不仅能正确预测训练集化合物的分子活性,还应当具有正确预测训练集之外其它分子生物活性的能力。因此,本研究使用66个训练集分子之外的SHP2催化位点抑制剂组成测试集,用于测试药效团正确预测外部分子活性的能力。图3显示了5个药效团模型对16个训练集化合物和66个测试集化合物预测活性值与实验活性值的回归分析结果。由图可见,5个药效团模型对训练集化合物的活性预测是非常准确的,化合物预测活性值和实验活性值间的相关系数在0.923 3~0.952 4。通过对5个药效团模型对测试集化合物预测活性值和实验活性值进行回归分析,得出药效团模型准确预测训练集之外化合物活性值的能力,从图3可见,5个药效团模型对66个测试集化合物的活性预测值和实验活性值之间的相关系数在0.822 8~0.909 1,其中P3和P5的预测准确度最高,相关系数分别为0.899 8和0.909 1。通过对化合物活性预测值和实验值相关系数的分析,本研究倾向选择P3和P5进行后续的虚拟筛选,以发现新型SHP2催化位点抑制剂。
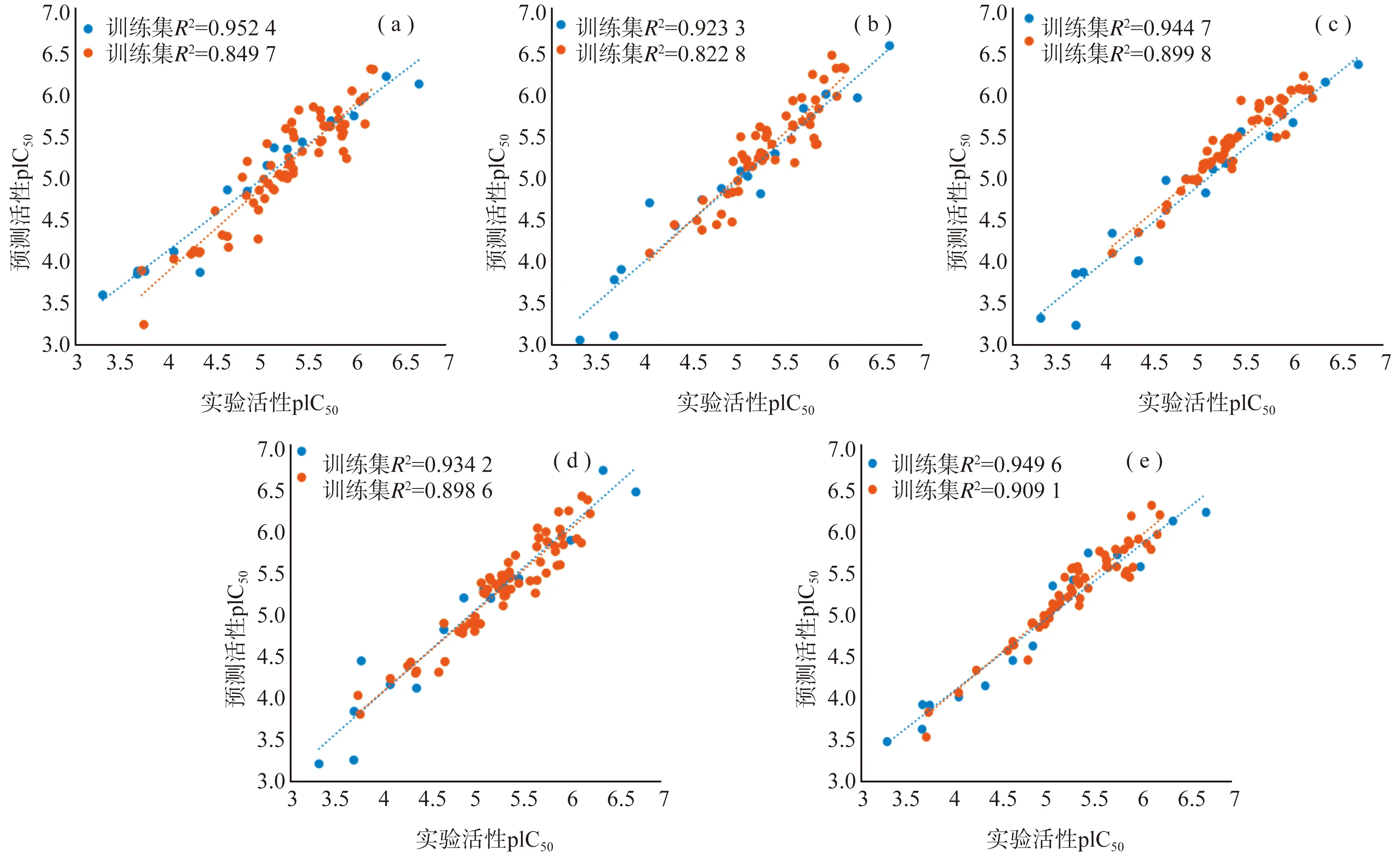
图3 5个药效团模型对训练集和测试集化合物活性预测值和实验值回归分析结果
除了测试集方法,本研究采用Fisher验证法对药效团进行随机交叉验证分析。在本次验证中,置信水平设为95%,程序将训练集化合物结构和活性值随机混乱组合,产生19个输入表单,这些表单参数与构建药效团模型时使用的参数设置完全相同,程序将产生95个药效团型。图4显示了新建的95个药效团模型和原始表单产生的5个药效团模型的Total cost值,比较发现原始表单产生的5个药效团模型除药效团P2外的Total cost值比随机表单产生的95个药效团的Total cost值低的多,这说明构建的药效团模型P1、P3、P4和P5具有很强的可信性。
为了进一步分析药效团模型P1、P3、P4和P5的有效性,将这4个药效团模型与训练集活性最高和活性最低的2个化合物进行匹配叠合。图5显示了定量药效团模型P1、P3、P4和P5与训练集中活性最高的化合物1(IC50= 0.2 μmol/L)和活性最低化合物16(IC50= 500 μmol/L)的匹配情况,从图5(a-d)可见,各药效团和化合物1匹配得非常完美,其每个药效特征元素均与化合物1中的相应化学功能基团匹配,而在图5(e-h)中药效团模型只与化合物16有部分匹配。因此,这些结果在一定程度上反映了各药效团的有效性。
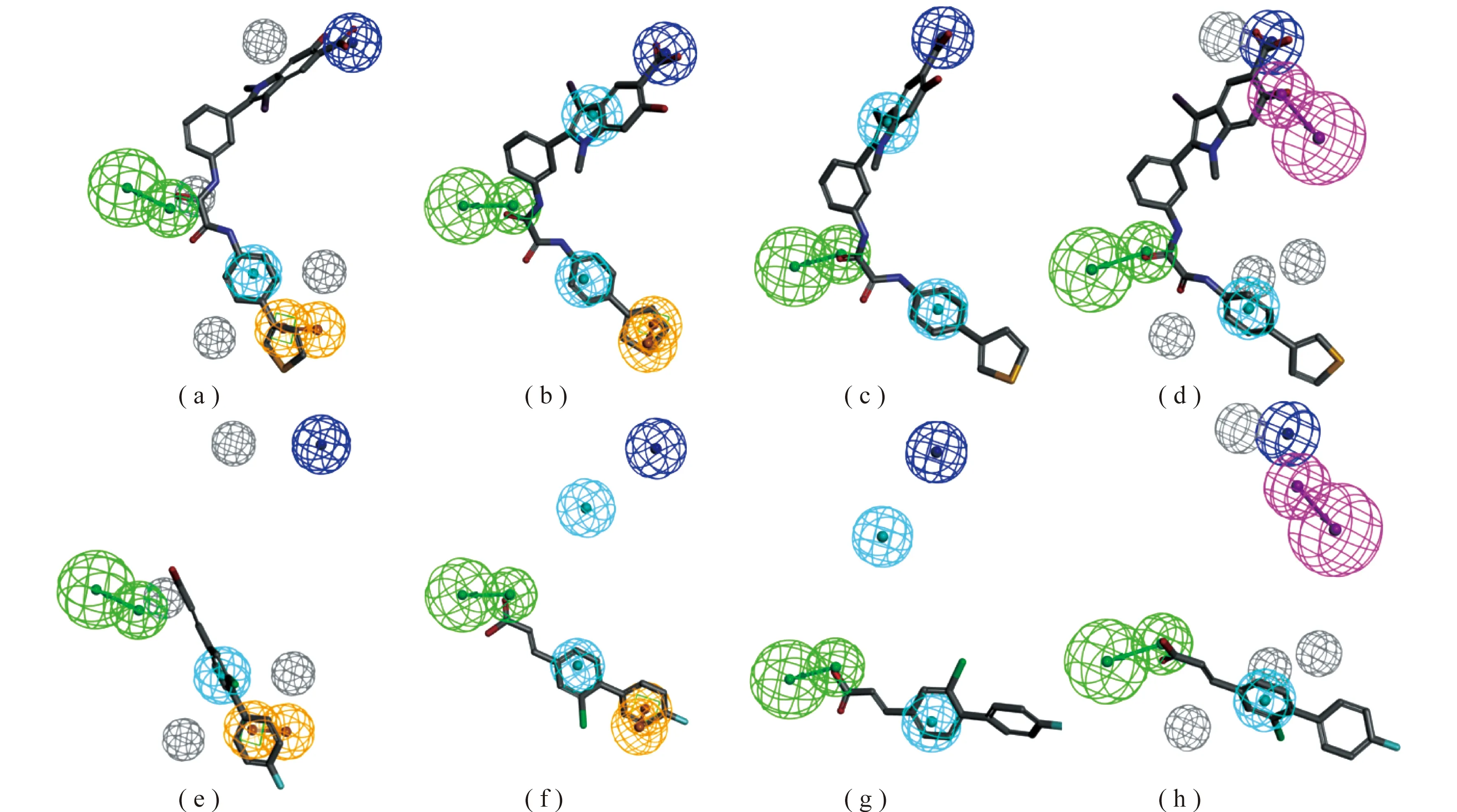
图5 药效团和训练集活性最高的化合物1(IC50:0.2 μmol/L)和活性最低化合物16(IC50:500 μmol/L)的匹配效果图(a~d)P1、P3、P4和P5与活性最高的化合物1的匹配图;(e~h)P1、P3、P4和P5与活性最低的化合物16的匹配图(绿色:氢键受体;玫红色:氢键供体;蓝色:疏水特征;黄色:芳香环特征;灰色:排斥球)
根据药效团模型对训练集和测试集化合物的活性预测能力,Fisher验证法分析,药效特征元素的组成和空间排布,及药效团和训练集化合物的匹配情况,最终选择药效团P3和P5进行后续的虚拟筛选。
3.3 分子对接参数和评分函数的确定
由于分子对接参数和评分函数对最终对接结果具有很大影响,因此在进行基于分子对接的虚拟筛选之前需要确定最优对接参数和评分函数。本研究选择SHP2催化结构域结合1个头孢磺啶衍生物的晶体复合物结构(PDB ID:4RDD)作为对接研究的受体,因为在所有已结晶SHP2催化结构域与催化位点抑制剂的复合物结构中,4RDD具有高分辨率(0.16 nm)。为了确定最佳的对接参数和评分函数,将已经与SHP2催化位点共结晶的4种活性化合物重新对接到SHP2的活性位点,调整对接参数和评分函数,直到小分子化合物对接构象尽可能与原来的结晶构象重合。经过不断测试,对接参数设置如:Fitness Function选用Chemscore,GA Parameters选用GOLD Default,Generate Diverse Solution设为True,Early Termination 设为False,其它参数保留软件默认参数。选用参数和评分函数的组合,使配体的对接构象与它们在晶体结构中的相应结合构象之间有最小均方根偏差(RMSD)值。表3列出了计算的RMSD值。4个化合物中3LU具有最小RMSD值0.186 95 nm,此值可接受。其它3个化合物的RMSD值都高于0.5 nm,这是因为这3个化合物本身柔性较大,与SHP2催化位点结合不够紧密,化合物三分之二的结构暴露于溶剂中与受体蛋白无任何相互作用,故对接构象与原结晶构象差别较大。基于化合物3LU的对接结果及对接软件GOLD5.1 在其它研究中的可靠性[29],表明GOLD软件在一定程度上能够重现配体的正确结合构象,可用于本研究中的分子对接研究。

表3 SHP2催化位点抑制剂的结晶构象和对接构象之间RMSD值
3.4 新型SHP2催化位点抑制剂的虚拟筛选
本研究采用3种计算机辅助药物发现技术,即贝叶斯分类模型,药效团模型和分子对接技术,进行多步骤的组合虚拟筛选以发现潜在新型SHP2活性位点抑制剂。虚拟筛选工作流程如图6所示。本研究所用3种计算机辅助药物发现技术中,基于贝叶斯分类模型的虚拟筛选速度最快,所以首先利用贝叶斯分类模型对包含129 087个化合物的大型商业化合物数据库进行SHP2催化位点抑制剂的预测;把预测为阳性的48 507个化合物构建为可用药效团模型进行筛选的3D数据库,利用比分子对接速度快很多的药效团模型进行3D数据库筛选;将筛选化合物与药效团匹配值高于4(FitValue 大于4.0)的158个化合物进行最后的分子对接筛选,根据对接化合物的打分值高低和观察对接构象与活性位点关键氨基酸残基的相互作用,最终选取了8个化合物进行购买和后续的酶水平活性验证。图7显示8个购买化合物的分子结构,表4列出8个化合物的筛选参数。
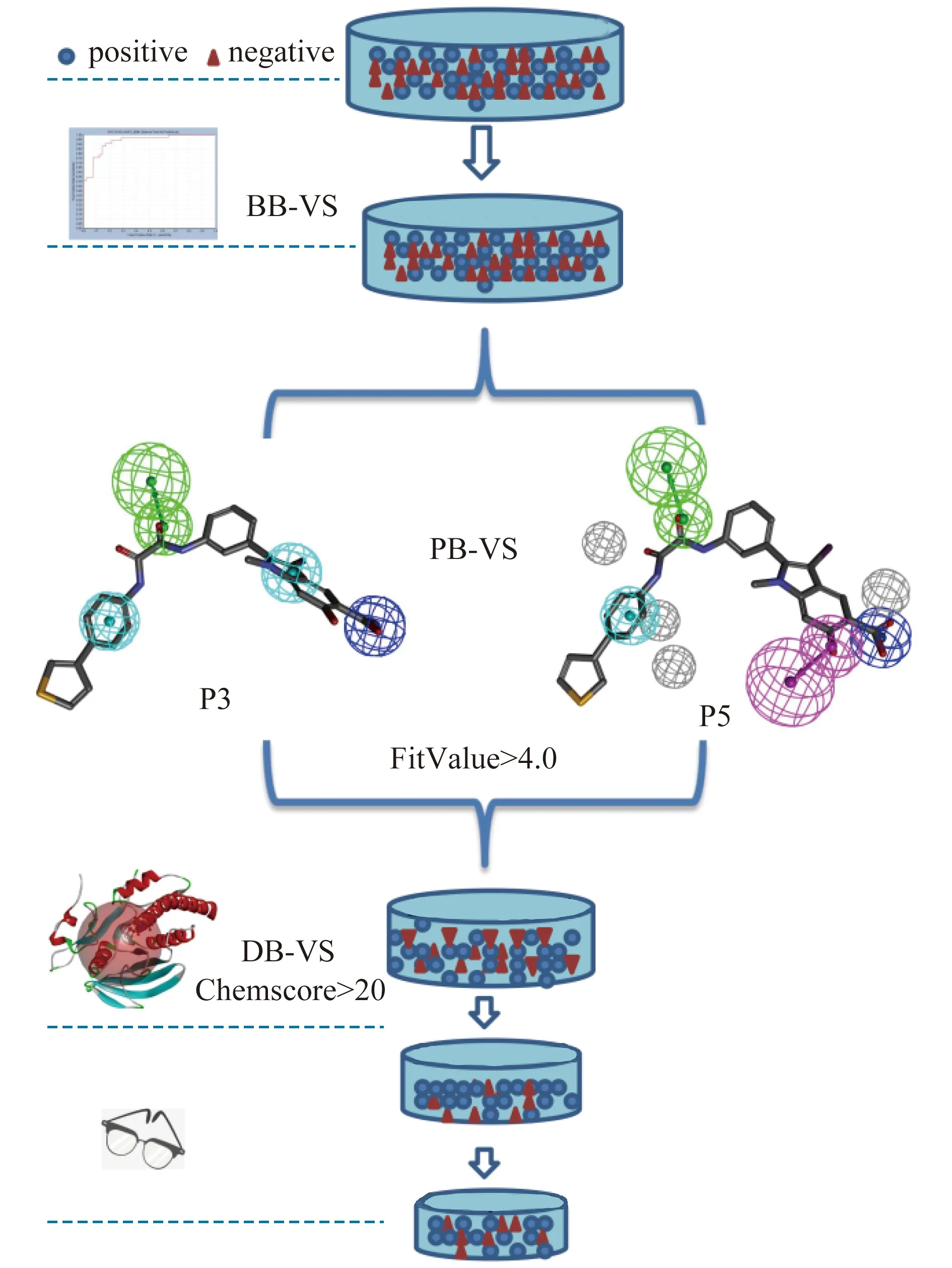
图6 本研究的虚拟筛选流程

图7 8个命中化合物的化学结构
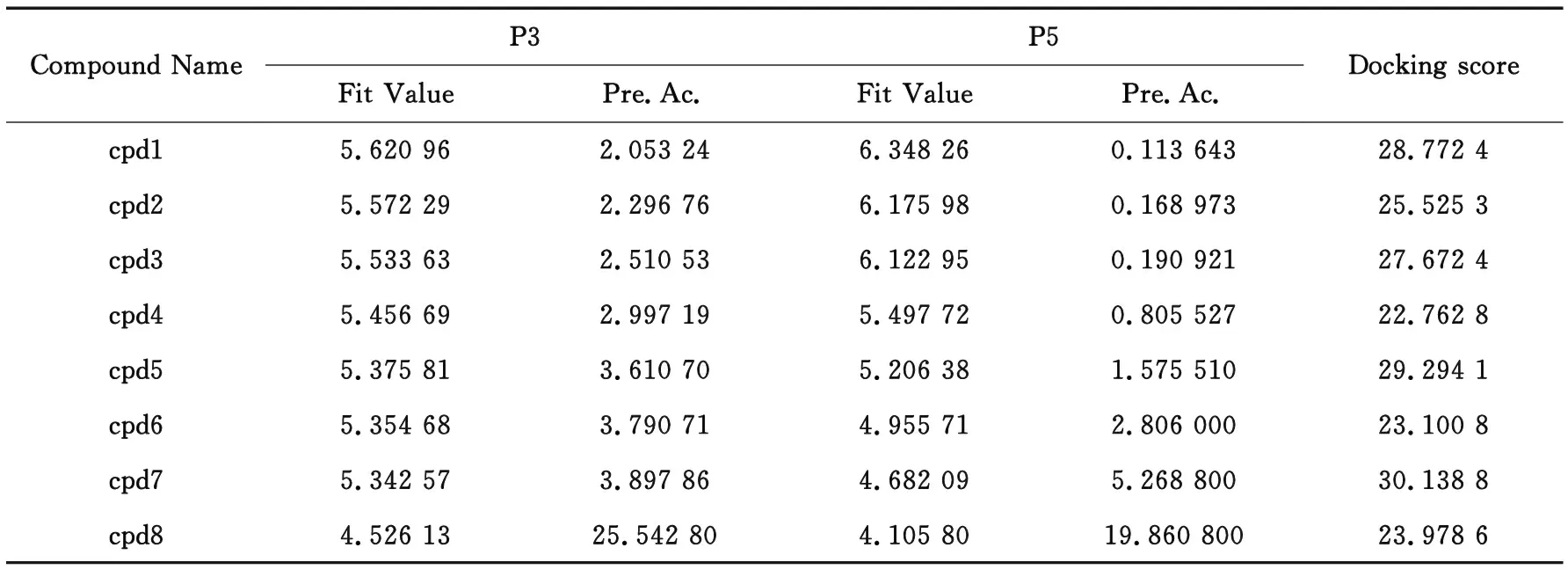
表4 8个命中化合物的筛选参数,包括药效团匹配值、3D-QSAR药效团模型预测的活性值和对接打分
3.5 新型SHP2催化位点抑制剂活性检测
化合物由LifeChemical化合物数据库国内代理商上海陶术购买(根据化合物售卖公司提供的材料显示,所有化合物纯度均大于等于90%),每种化合物皆用DMSO溶解,精确配置成10 mmol/L的母液,每个待测化合物取20 μL母液,寄往英国Eurofins Discovery公司完成酶水平活性检测。图8显示化合物在10 μmol/L浓度下对SHP2活性的抑制率,化合物cpd2、cpd4和cpd6在浓度为10 μmol/L时对SHP2催化活性具有明显抑制能力,抑制率分别为79%,71%和67%。为了进一步检测活性最高化合物抑制不同磷酸酶催化活性的选择性,检测了cpd2 在10 μmol/L浓度时,对SHP1、PTP1B和DUSP22催化活性的抑制。如图9所示,化合物cpd2 在10 μmol/L浓度时,对SHP1、PTP1B和DUSP22的酶活抑制率分别为42%,41%和34%,与同浓度cpd2对SHP2酶活抑制率21%相比较,提示化合物cpd2具有一定潜在选择性。酶活检测表明,化合物cpd2是一个具有潜在选择性的SHP2催化位点抑制剂,具有进一步改构的价值。
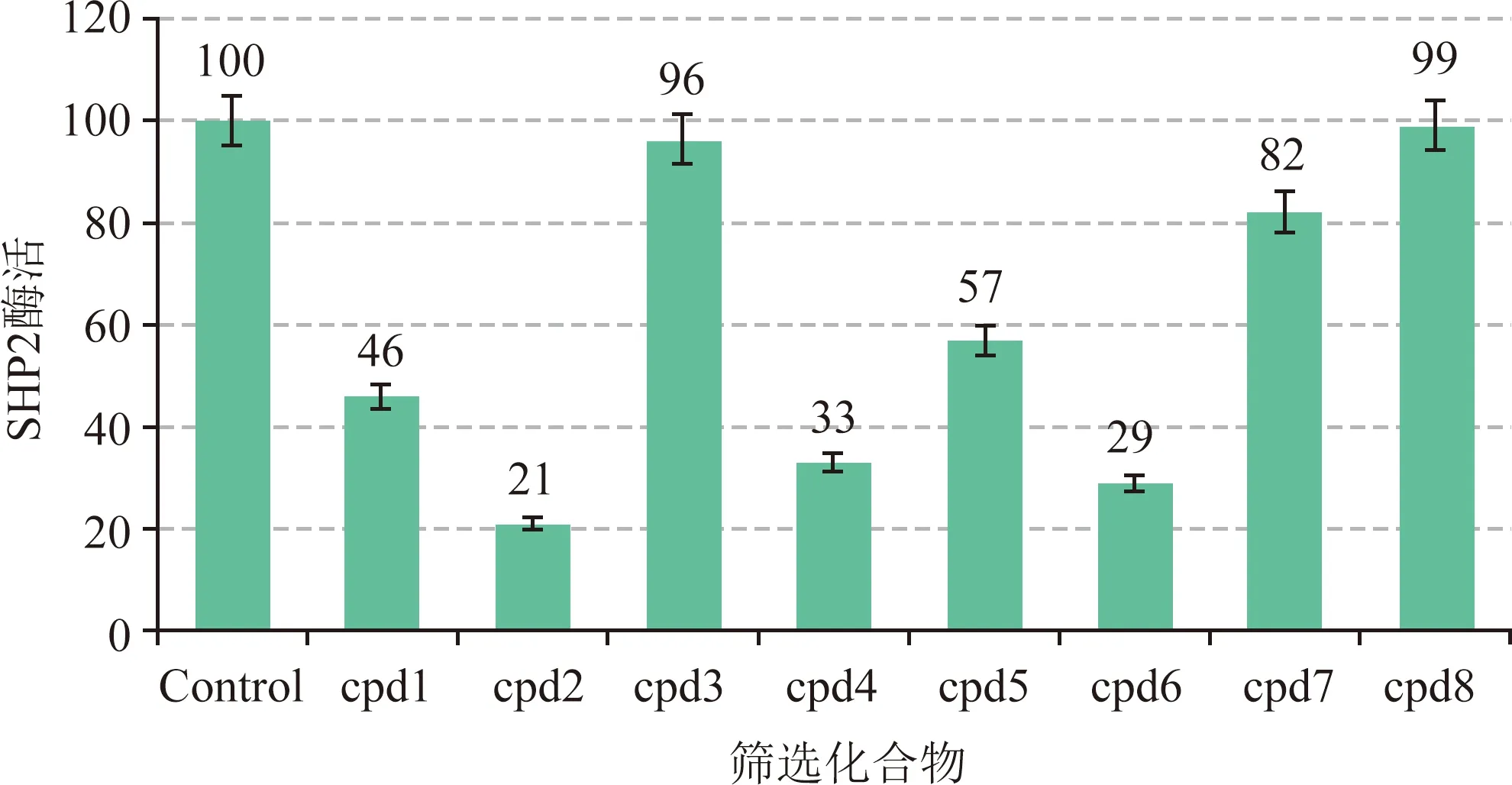
图8 8个筛选命中化合物抑制SHP2酶活性结果
3.6 活性化合物cpd2与药效团模型的匹配及分子对接结果分析
为了更好地解释化合物cpd2结构和活性间的关系,分析了该化合物和药效团的匹配情况,及其与SHP2催化活性位点的对接结果,以阐明化合物cpd2为何会抑制SHP2催化活性。
从图10(a)中可以看到,命中化合物cpd2与药效团模型P3匹配效果较好。具体地说,2个疏水特征与cpd2的苯基匹配,解离负电荷特征与羧基匹配,氢键受体与硫氧代噻唑啉上的氧原子匹配,芳香环特征没有匹配到化合物cpd2的结构上。相比较,化合物cpd2与药效团模型P5匹配效果好于其与P3的匹配效果,如图10(b)。具体来说,解离负电荷特征与羧基匹配,氢键受体与硫氧代噻唑啉上的氧原子匹配,氢键受体与羟基匹配,疏水特征与甲氧基匹配。图10(c)显示了cpd2在SHP2催化活性位点中可能的结合构象,水杨酸结合在SHP2底物磷酸化部位的结合位点,其中羧基与GLN510和GLY464形成氢键相互作用,羟基和CYS459形成氢键相互作用,苯环与ALA461形成疏水相互作用并与LYS366形成正电荷-π相互作用;硫氧代噻唑啉上的氧原子与SER460形成氢键相互作用,硫氧代噻唑啉的五元环与LYS364形成疏水相互作用;邻二甲氧基苯上的一个甲氧基与ARG278形成弱氢键相互作用。化合物cpd2与药效团的匹配结果与其和SHP2催化活性位点结合形成的相互作用相吻合,说明药效团预测的cpd2活性构象和分子对接预测的活性构象基本吻合,化合物cpd2极可能是采取此种构象结合在SHP2催化位点抑制其催化活性。药效团匹配和分子对接的结果阐明了化合物cpd2抑制SHP2催化活性的作用机制。
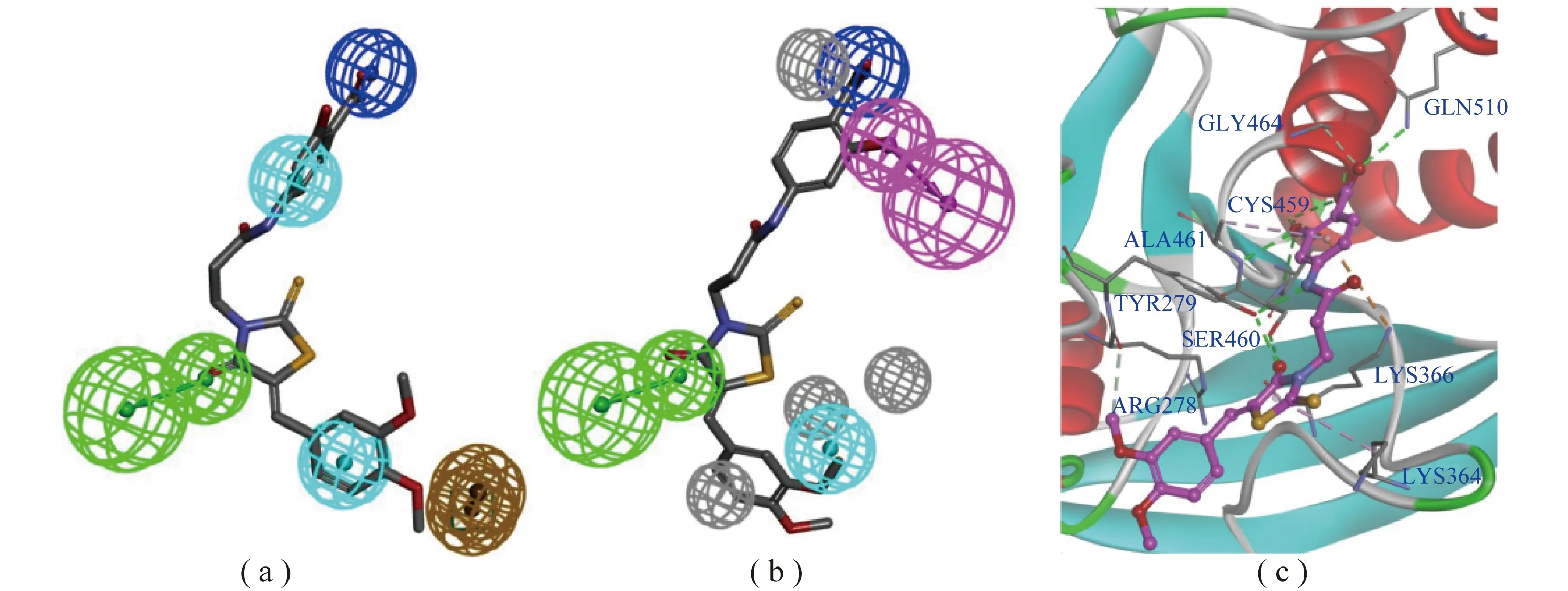
图10 (a)和(b)分别是药效团P3和P5与化合物cpd2的匹配结果(绿色:氢键受体,玫红色:氢键供体,蓝色:疏水特征,黄色:芳香环特征,灰色:排斥球);(c)cpd2在SHP2催化活性位点中可能的结合构象(绿色:氢键,浅粉:疏水相互作用,橙色:静电相互作用,灰绿:弱氢键)
4 结论
本研究利用计算机辅助药物设计技术结合酶水平活性测试,发现了3个新型SHP2催化位点抑制剂,其中活性最高化合物cpd2具有一定的潜在选择性。通过分析cpd2与SHP2催化位点抑制剂3D-QSAR药效团的匹配结果以及cpd2与SHP2催化活性位点的对接结果,发现药效团预测的活性构象与分子对接预测的活性构象相当吻合,且cpd2与药效团关键药效特征的匹配与其和SHP2催化活性位点关键氨基酸残基的相互作用基本一致,所以阐明了cpd2抑制SHP2酪氨酸磷酸酶活性的作用机制。
猜你喜欢
故事大王(2017年11期)2018-01-21
温州大学学报(自然科学版)(2016年1期)2016-10-27
新疆农垦科技(2016年2期)2016-08-21
中学化学(2015年12期)2016-01-19
应用化工(2014年7期)2014-08-09
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年5期)2014-02-28
无机化学学报(2014年1期)2014-02-28
食品科学(2013年13期)2013-03-11
物理化学学报(2012年12期)2012-03-06
