
铁死亡在骨代谢功能细胞中的机制与展望
2024-01-12 13:13秦金然王亮王礼宁朱三木马勇谢雁鸣魏戌章轶立
中国骨质疏松杂志 2023年12期
秦金然 王亮 王礼宁,2 朱三木 马勇,2 谢雁鸣 魏戌 章轶立,2*
1.南京中医药大学中西医结合学院,江苏 南京 210023 2.江苏省中医退行性骨关节病临床医学创新中心,江苏 无锡 214071 3.中国中医科学院中医临床基础医学研究所,北京 100700 4.中国中医科学院望京医院,北京 100102
骨骼是人体中代谢相对活跃的组织,其正常生理功能与骨细胞、骨髓间充质干细胞、成骨细胞、破骨细胞等功能细胞的正常运转有关。近年来,以骨质疏松症为代表的骨代谢疾病发病率呈现逐年上升趋势[1]。2018年国家卫生健康委发布的我国骨质疏松症流行病学调查结果显示,我国50岁以上人群的骨质疏松症患病率为19.2%,65岁以上人群的骨质疏松症患病率高达32.0%。随着我国老龄人口数量的日益增加,骨质疏松症患病率亦呈急速攀升趋势,已成为政府、社会、个人亟待重视的公共健康问题[2-3]。
“铁死亡”是由Brent R.Stockwell教授等于2012年首次提出,以细胞内活性氧(reactive oxidative species,ROS)堆积以及线粒体形态、膜电位改变为主要特征[4-5]。目前,关于铁死亡的研究主要集中在肿瘤、神经退行性疾病、视网膜疾病、自身免疫性疾病等领域[6]。近年来,铁死亡与骨质疏松症为代表的骨代谢疾病研究取得了诸多进展。因此,本综述拟从骨细胞、骨髓间充质干细胞、成骨细胞、破骨细胞等骨代谢功能细胞的角度,重点阐述铁死亡发生机制及其与相关细胞的作用关系。
1 铁死亡概述
与传统的程序性细胞死亡(如细胞凋亡、坏死)方式不同,铁死亡主要归因于细胞内脂质过氧化、谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)抗氧化系统失调以及铁超负荷[7-8]。在形态学上,铁死亡涉及细胞膜破裂和出泡,线粒体变小、膜密度增加、线粒体嵴减少甚至消失、线粒体外膜破裂,细胞核大小正常但缺少染色质凝聚[9]。
1.1 脂质过氧化
GPX4是铁死亡的关键调节因子,其通过将高反应性脂质氢过氧化物还原为非反应性脂质醇来保护细胞免受膜脂质过氧化并维持氧化还原稳态[10]。
根据烃链饱和度的差异,脂肪酸可分为三类:饱和脂肪酸(saturated fatty acids,SFAs)、单不饱和脂肪酸(monounsaturated fatty acids,MUFAs)和多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)[11]。含有PUFAs的磷脂极易发生过氧化,促进 PUFAs 掺入膜磷脂可以促进铁死亡;反之,增加膜磷脂中氧化性较低的MUFAs数量可以有效抵抗铁死亡[12]。因此,控制PUFAs和MUFAs代谢的酶和通路在确定细胞对铁死亡敏感性方面起关键作用。Mortensen等[13]认为PUFAs在铁死亡过程中占据重要地位,因为它们倾向于形成过氧自由基,通过链式反应在整个膜中传播,继而导致不可挽回的膜损伤和细胞死亡。而脂氧合酶(lipoxygenases,LOXs)和磷酸化酶激酶G2(phosphorylase kinase gamma 2,PHKG2)是铁死亡过程中脂质过氧化的两个关键驱动因素。脂氧合酶的过表达使细胞对铁死亡敏感[14]。PHKG2可调节脂氧合酶对铁的可利用性,并且脂氧合酶通过双烯丙基位置的多不饱和脂肪酸过氧化促使铁死亡[15]。此外,相关学者还发现脂氧合酶通过PHKG2依赖性铁池氧化PUFAs是铁死亡所必需的,并且GPX4中催化硒化半胱氨酸的共价抑制阻止了PUFAs氢过氧化物的消除,这些发现为在不同情况下调控铁死亡提供了新的研究思路[16]。
1.2 GPX4抗氧化系统失调
氧化应激引起不受控制的脂质过氧化物产生通常会导致线粒体脂质过氧化和损伤,继而引发铁死亡。脂质过氧化物是脂质过氧化过程中的关键中间体,脂质氢过氧化物酶GPX4将脂质氢过氧化物蜕变为脂质醇,此过程可防止铁(Fe2+)依赖性形成有毒脂质ROS。GPX4活性的丢失及随后脂质氢过氧化物的积累会导致铁死亡,既往研究表示GPX4是铁死亡发生过程中的中枢调节因子,其可以在复杂的细胞膜环境中加速脂质过氧化物的衰减[17]。
铁死亡诱导因子可通过多种途径直接或间接的影响谷胱甘肽过氧化物酶(glutathione peroxidase,GPX),导致抗氧化能力下降和细胞中脂质活性氧积累,最终导致氧化细胞死亡。erastin和RSL3是最早的GPX系统抑制剂,具有不同的细胞死亡触发机制。erastin通过抑制系统XC-阻止胱氨酸的摄取,从而消耗谷胱甘肽(glutathione,GSH),发挥间接抑制GPX4的作用;而RSL3则是直接灭活GPX4,诱导铁死亡[18]。与之前描述的铁死亡诱导剂不同,Gaschler等[19]研究发现FINO2可启动多管齐下的铁死亡诱导机制。Shimada等[20]发现CIL56和FIN56催化的细胞死亡常常伴随着脂质ROS的产生,且维生素E和铁螯合剂对这些化合物诱导的细胞死亡具有抑制作用,表明它们可能是铁死亡诱导剂。此外,作者还证实了 FIN56通过诱导GPX4降解来诱导铁死亡。
1.3 铁代谢紊乱
铁离子对于细胞生长、氧气利用以及各种酶的活性等诸多的细胞和分子过程至关重要,反之,过量的铁离子会通过推动有毒自由基的产生继而致使细胞功能障碍,异常的铁积累可能会导致多种器官损伤[21]。细胞铁超负荷可降低细胞活力、超氧化物歧化酶和GSH水平,增加了ROS产生、脂质过氧化、丙二醛水平以及铁死亡相关蛋白的表达,并诱导线粒体的超微结构变化[22]。大量Fe3+在金属还原酶的作用下还原成Fe2+,游离Fe2+氧化性强,易于与H2O2发生芬顿反应,产生羟基自由基,继而促进脂质过氧化反应发生损伤细胞膜致使细胞死亡[23]。
Geng等[24]通过体外实验证明敲低膜铁转运蛋白(ferroportin,FPN)会增加铁依赖性脂质ROS积累继而加速erastin诱导的铁死亡。后续的相关研究再次验证了这一结果,并提示FPN过表达会抑制erastin诱导的铁死亡[25]。
2 铁死亡与骨代谢功能细胞
2.1 铁死亡与骨细胞
一直以来,骨细胞被认为是成骨细胞和破骨细胞传递调节信号的“指挥官”,在控制骨重塑中发挥关键作用。Ma等[26]认为骨细胞是预防和治疗铁超负荷引起的骨质疏松症的主要靶点,在指定浓度的柠檬酸铁胺(ferric ammonium citrate,FAC)处理24 h后,FAC以剂量依赖的方式显著增加骨硬化蛋白和RANKL的表达水平,同时降低OPG的表达水平,导致RANKL/OPG的表达水平升高,而使用去铁胺(Deferoxamine,DFO)和N-乙酰半胱氨酸(N-acetylcysteine,NAC)在处理FAC干预的骨细胞可逆转该结果。研究发现铁超负荷不仅会破坏骨细胞微丝骨架,并且会导致骨细胞中微丝骨架的分布改变;此外,铁超负荷显著阻碍MLO-Y4细胞系的细胞活力,同时通过上调骨细胞中RANKL的表达和分泌刺激破骨细胞分化[27]。糖尿病微环境可显著促进骨细胞铁死亡,主要表现为大量的脂质过氧化、铁超负荷以及铁死亡途径的异常激活。靶向铁死亡(注射铁死亡抑制剂Fer-1)或 HO-1(注射HO-1抑制剂ZnPP)通过破坏脂质过氧化和HO-1激活之间的恶性循环,有效减少糖尿病骨质疏松症(diabetic osteoporosis,DOP)中的骨细胞死亡,最终减少骨质流失[28]。Sun等[29]的研究结果表明地塞米松(DEX)可上调p53以抑制溶质载体家族7成员11(solute carrier family 7 member 11,SLC7A11)/GPX4的表达,从而诱导MLO-Y4细胞(小鼠骨样细胞)铁死亡。
2.2 铁死亡与骨髓间充质干细胞
骨髓间充质干细胞(bone mesenchymal stem cells,BMSCs)是一种多能干细胞,其在正常骨代谢中起关键作用,被认为是具有维持和修复骨组织潜力的种子细胞[30-31]。近年来,随着对铁死亡研究的深入,人们发现可通过PI3K/AKT/mTOR信号通路抑制铁死亡来改善BMSCs的抗氧化应激过程[32]。Li等[33]的实验结果显示地塞米松诱导BMSCs铁死亡,且褪黑素可通过PI3K/AKT/mTOR途径减少地塞米松诱导的BMSCs铁死亡。
Engeletin(二氢山奈酚 3-鼠李糖苷)是一种从水果和蔬菜中分离出来的天然化合物[34]。研究发现Engeletin对BMSCs的成骨能力具有积极影响,此外还可以通过Nrf2/Keap1途径减轻erastin诱导的氧化应激损伤,抑制BMSCs铁死亡[35]。Liu等[36]使用erastin诱导BMSC铁死亡,发现并首次证明了NOP2/Sun RNA甲基转移酶5(NOP2/Sun RNA methyltransferase 5,NSUN5)通过与肿瘤坏死因子受体相关蛋白(tumor necrosis factor receptor associated protein 1,TRAP1)相互作用以及铁蛋白重链1(ferritin heavy chain 1,FTH1)和铁蛋白轻链(ferritin light chain,FTL)mRNA的5-甲基胞嘧啶(5mC)修饰来抑制BMSCs的铁死亡。由此可见,NSUN5-FTH1/FTL通路组分的治疗靶点具有提高BMSCs存活的潜力。
Song等[37]发现,FA互补基团D2(FA complementation group D2,FANCD2)可以通过减少铁超负荷和脂质过氧化,抑制erastin刺激的BMSCs铁死亡。并且ROS的过度积累也会激活G蛋白轴并破坏氧化平衡稳态以诱导BMSCs中的铁死亡。Perillo等[38]认为p53通过在中度氧化应激下阻止ROS的过度增加来促进细胞存活,而当氧气增加超过阈值水平时,它则会转变为ROS诱导剂,触发细胞死亡。高糖环境中,BMSCs的成骨细胞分化与细胞增殖活性被显著抑制,致使细胞内ROS和LPO水平增高,而铁死亡抑制剂Fer-1可恢复其正常[39]。除此之外,已有研究表明由FAC引起的细胞内铁超负荷可以通过下调Wnt靶基因Lef1、Bmp4、Smad6 和细胞周期蛋白D1(Cyclin D1)的表达来提高铁死亡敏感性,从而抑制间充质干细胞向成骨细胞分化[40]。最后,维生素K2(VK2)也已被证实可通过腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)/SIRT1信号通路抑制BMSC铁死亡,从而缓解2型糖尿病骨质疏松症(type 2 diabetes osteoporosis,T2DOP)[41]。
2.3 铁死亡与成骨细胞
成骨细胞是BMSCs的终末分化产物,调节骨的形成以及重建过程。铁是众多生物过程的必需微量元素,在成骨细胞的分化和矿化过程中起关键作用。在相似的条件下,为实现适当的分化和矿化,成骨细胞比脂肪细胞有更高的铁需求[42],但铁超负荷同样会抑制成骨作用,导致成骨细胞铁死亡[43]。
铁转运蛋白(ferroportin1,FPN1)是目前发现的哺乳动物中唯一的细胞中铁外排蛋白,是铁死亡信号通路的关键蛋白[44-45]。FPN1通过提高细胞内ROS水平,引起氧化应激,从而导致细胞铁死亡。付殷等[46]的研究表明,淫羊藿苷可以通过抑制FPN1过表达调节铁死亡信号通路,最终提高成骨细胞增殖、矿化能力。根据前期研究可知,AKT/PI3K信号通路是miR-483-5p-SATB2轴在OVX大鼠中作用的下游靶标,通过促进骨髓干细胞的增殖和迁移改善骨质疏松症的发展[47]。Hao等[48]验证了ATM和AKT/PI3K通路可改善成骨功能并抑制成骨细胞铁死亡。此外,Luo等[40]在机制上揭示了铁剂量依赖性抑制Wnt信号传导。Wnt 激动剂、铁死亡抑制剂或抗氧化剂褪黑激素可逆转铁被抑制的Wnt信号传导,通过减少ROS和脂质过氧化物(lipid peroxidation,LPO)的产生恢复成骨细胞分化,从而在不降低铁超负荷的情况下显著预防铁死亡。另有研究证实,DEX通过p53/SLC7A11/GPX4通路诱导MC3T3-E1细胞铁死亡[29]。
高糖可通过积累RAS以及消耗细胞内的GSH来诱导MC3T3-E1成骨细胞的铁死亡[49]。Zhao等[50]的研究表明高糖可诱导转录激活因子3(activation transcription factor 3,ATF3)上调,通过抑制ATF3的功能可增加GPX4水平并减少ROS和脂质过氧化物的积累,从而达到抑制成骨细胞铁死亡的目的。线粒体铁蛋白(mitochondrial ferritin,FtMt)的过表达可降低高糖条件下成骨细胞发生铁死亡,并且FtMt下调可通过ROS/PTEN诱导假定激酶1(PTEN-induced putative kinase 1,PINK1)/帕金森病蛋白(parkinsons disease protein,Parkin)通路诱导线粒体自噬,促进成骨细胞铁死亡[49]。晚期糖基化终产物(advanced glycation end products,AGEs)是在高糖条件下形成的一组具有破坏潜力的修饰蛋白质和(或)脂质[51]。Ge等[52]在研究中首次揭示AGEs通过诱导铁死亡显著抑制了成骨细胞的增殖、分化和矿化。研究表明高脂肪环境同样会抑制成骨细胞增殖和成骨分化,铁死亡的相关指标随之发生显著变化,并且发现铁死亡抑制剂可抑制由于高脂肪饮食导致的小鼠骨质流失的发展[53]。MaR1(Maresin1)是一种由多不饱和脂肪酸产生的内源性促消退脂质介质,其在体内外通过激活核因子E2相关因子2(nuclear factor erythroid 2-related factor 2,Nrf2)抑制高葡萄糖诱导的成骨细胞铁死亡,缓解T2DOP[54]。激活METTL3/凋亡信号调节激酶1(apoptosis signal-regulating kinase 1,ASK1)-p38信号通路可能是高糖高脂肪诱导成骨细胞铁死亡继而导致糖尿病骨质疏松症的主要原因之一[55]。ASK1-p38信号通路是一个与铁离子浓度密切相关的信号转导途径,研究发现铁超负荷可以通过调控ASK1-p38信号通路调控成骨细胞铁死亡[56]。最后,研究发现褪黑素可通过激活Nrf2/HO-1信号通路,显著降低MC3T3-E1的铁死亡水平,提高成骨能力[57]。
2.4 铁死亡与破骨细胞
破骨细胞是体内唯一具有骨吸收能力的细胞。据报道,骨髓来源巨噬细胞(bone marrow derived macrophage,BMM)中产生的ROS会增强RANKL诱导破骨细胞分化的能力[58]。有研究表明,过量的铁进入细胞内会通过芬顿反应产生ROS,并进一步激活细胞内丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路。ROS/MAPKs/核转录因子-κB (nuclear factor-kB,NF-kB)/核苷酸结合寡聚化结构域样受体蛋白3(nod-like receptor protein 3,NLRP3)的激活,可促进糖尿病性骨质疏松中破骨细胞介导的骨丢失[59]。Ni等[60]研究也证实了铁死亡参与RANKL诱导的破骨细胞分化,并且在该过程中铁饥饿反应和正常氧浓度下的铁蛋白吞噬有助于RANKL诱导的破骨细胞铁死亡;此外,作者还证明了体内HIF-1α可抑制通过铁蛋白吞噬诱导的铁死亡发生。Qu等[61]使用RANKL诱导的细胞模型评估唑来膦酸(ZA)对破骨细胞功能的调节作用,结果显示铁离子、ROS以及MDA含量增加,GPX4和GSH水平降低,由此可见,ZA处理抑制了破骨细胞的细胞活力,促进破骨细胞铁死亡。
铁调素是一种富含半胱氨酸的小分子肽,其由肝细胞产生,可抑制FPN活性[62],是铁代谢和铁稳态的关键调节剂,它受到铁超负荷和炎症的刺激,可以通过利用铁调素来增加铁的积累[63]。铁可以增加破骨细胞的脂质过氧化,并诱导铁死亡,从而抑制破骨细胞分化[64]。Li等[65]的研究认为过量的铁通过增加TNF-α的分泌来破坏小鼠的骨承重能力,且TNF-α会促进破骨细胞的分化并增强骨吸收。
在破骨细胞分化过程中,破骨细胞通过转铁蛋白受体1(transferrin receptor 1,TfR1)介导的转铁蛋白(transferrin,Tf)内吞作用吸收大量铁。Jin等[64]发现Tf能增强青蒿琥酯对破骨细胞的活力和分化的抑制作用,作者认为其机制与青蒿琥酯诱导破骨细胞铁死亡有关。
鉴于铁死亡在不同骨代谢功能细胞涉及的关键分子、信号通路、外部影响因素、组织局部微环境等不同,相关总结信息见表1、图1。
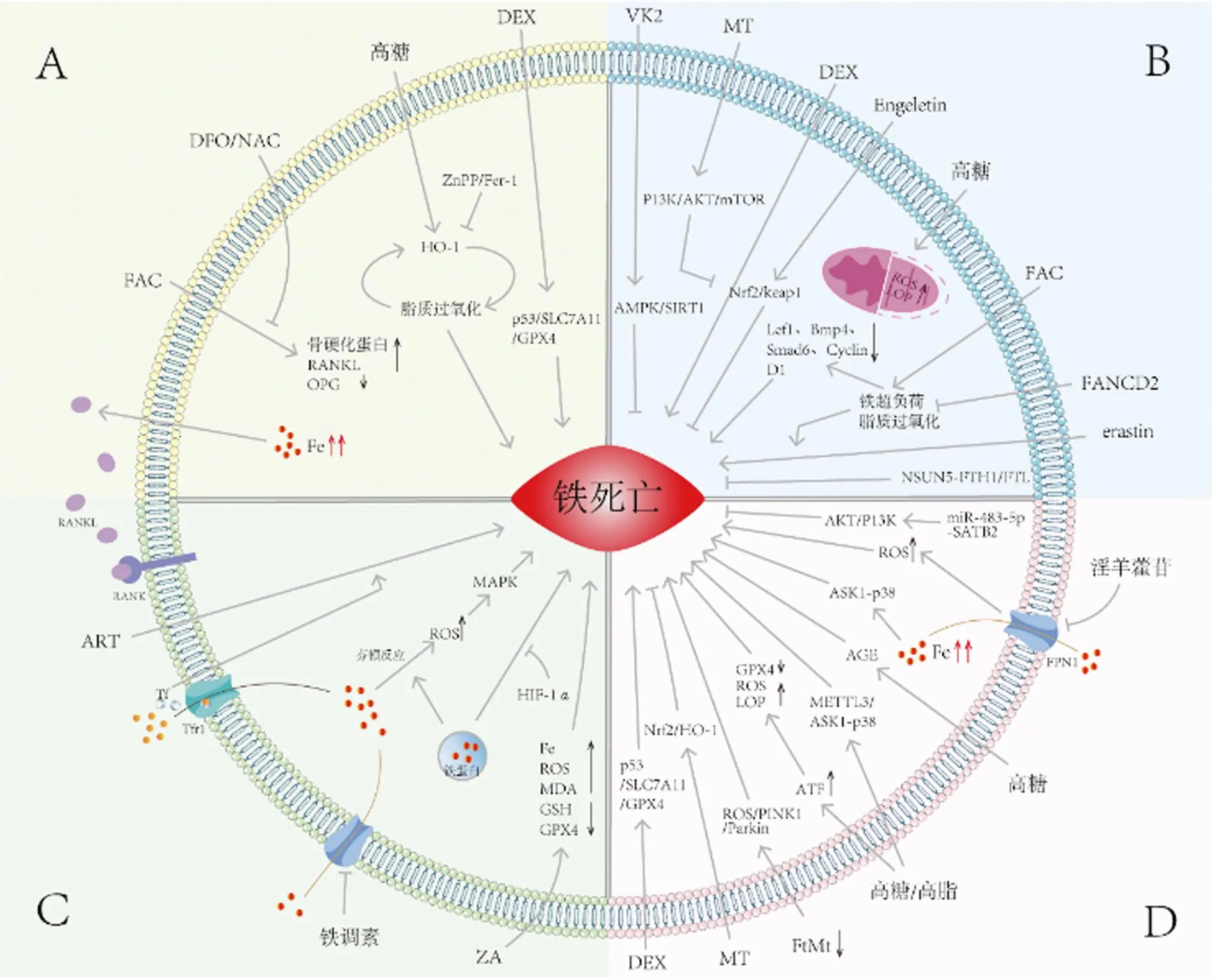
图1 铁死亡在骨代谢功能细胞中的作用机制Fig.1 Mechanism of action of ferroptosis in functional cells of bone metabolism注:A:骨细胞;B:骨髓间充质干细胞;C:破骨细胞;D:成骨细胞;ZnPP:HO-1抑制剂;Fer-1:铁死亡抑制剂;FAC:柠檬酸铁胺;DFO:去铁胺;NAC:N-乙酰半胱氨酸;SLC7A11:溶质载体家族7成员11;DEX:地塞米松;FtMt:线粒体铁蛋白;VK2:维生素K2;MT:褪黑素;Engeletin:二氢山奈酚 3-鼠李糖苷;FANCD2:FA互补基团D2;erastin:铁死亡诱导剂;AGE:晚期糖基化终产物;ROS:活性氧;LOP:脂质过氧化物;GPX4:谷胱甘肽过氧化物酶4;FPN1:铁转运蛋白1;PINK1:PTEN诱导假定激酶1;Parkin:帕金森病蛋白;MAPK:丝裂原活化蛋白激酶;ZA:唑来膦酸;GSH:谷胱甘肽;ART:青蒿琥酯;Tf:转铁蛋白;Tfr1:转铁蛋白受体1;MDA:丙二醛;RANK:核因子-κB受体活化体;RANKL:核因子-κB受体活化体配体;OPG:骨保护素。

表1 骨质疏松症相关细胞与铁死亡之间的联系Table 1 Association between osteoporosis-associated cells and ferroptosis
3 总结与展望
越来越多的证据表明,铁死亡参与骨代谢疾病的病理生理过程,通过抑制或刺激这种独特的细胞死亡方式来干预骨质疏松症为代表的骨代谢疾病成为未来研究的重点。因此,持续关注铁死亡在目标疾病发生发展中的作用,或可为开发新的治疗策略提供希望。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17
河北科技师范学院学报(2022年2期)2022-08-26
现代临床医学(2021年6期)2021-11-20
中国骨质疏松杂志(2021年9期)2021-10-08
中国临床医学(2019年3期)2019-01-04
中成药(2017年12期)2018-01-19
中国塑料(2016年3期)2016-06-15
中国骨质疏松杂志(2016年1期)2016-01-29
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
中国洗涤用品工业(2015年4期)2015-02-28
