
不同分型钙通道阻滞剂在吗啡成瘾中的作用
2011-05-29 12:42朱永平
中国药理学通报 2011年2期
张 乐,朱永平
(浙江大学医学院毒理研究室,浙江杭州 310058)
药物成瘾是一种复杂的慢性复发性脑病,其形成机制复杂,尚不完全清楚,一般认为与兴奋性氨基酸、Ca2+、一氧化氮(NO)和环鸟苷酸(cGMP)等有关。其中神经细胞内重要的第二信使Ca2+通过与阿片受体偶联,实现其跨膜信号的转导,参与镇痛和成瘾作用,其重要性日益受到人们的重视。
1 吗啡对细胞内Ca2+浓度的影响
大量研究证明,Ca2+在吗啡镇痛作用和成瘾形成的过程中发挥着重要作用,吗啡通过阿片受体影响Ca2+通道的活性,改变中枢和外周神经细胞内Ca2+浓度,Ca2+通道阻滞剂可治疗吗啡依赖动物的戒断症状。
吗啡急性作用时,通过激动μ阿片受体抑制电压依赖型Ca2+通道,使Ca2+内流减少,降低细胞内Ca2+浓度,影响感觉传导过程而产生抗伤害作用。长期给予吗啡,阿片受体脱敏,细胞内腺苷酸环化酶-环化磷酸腺苷(AC-cAMP)系统上调等变化,使μ阿片受体对Ca2+通道抑制作用减弱。一旦停用吗啡或用纳络酮激发戒断反应,则出现Ca2+内流增多,细胞内Ca2+浓度升高,使兴奋性神经递质释放增多,引起戒断反应。长期应用阿片类药物使神经细胞内Ca2+增加的途径主要有两个:①细胞外Ca2+经其通道内流入胞内;②细胞内钙库释放Ca2+。因此通过阻断Ca2+通道,抑制外钙内流和内钙释放是阿片类物质发挥镇痛作用延缓依赖形成的机制之一。
2 钙通道分类
钙通道位于细胞膜上,是由 α1、α2、β、γ、δ 五个亚基组成的多聚体蛋白质,中央围成的一个小孔通道。1955年Hodgkin提出“通道概念”,1949年Cole用膜片钳证实并于1963年获诺贝尔奖。目前已知的钙通道大致分为以下几型:①电压依赖型钙通道(voltage-dependent calcium channel,VDCC):L型、N型和T型等;② 激动剂—受体门控Ca2+通道(ligand-gated calcium channel,LGCC):ATP、N-甲基-D-天冬氨酸均可与相应受体结合,促进Ca2+内流;③ 机械操纵型Ca2+通道:此型仅见于血管内皮细胞;④ 质膜上分布的钙泵;⑤ IP3受体Ca2+通道;⑥ 雷诺定受体Ca2+通道(ryanodine,RYR钙通道)。前两型分布在细胞膜上,可通过抑制外钙内流降低胞浆钙浓度;最后两型分布在内质网和肌浆网上面,通过抑制内钙释放延缓吗啡依赖形成[1]。
3 钙通道调节剂对吗啡成瘾作用的影响
许多实验从整体、离体和分子水平探讨证明:吗啡急性作用阶段,突触和突触囊泡内钙浓度下降,此时钙通道激动剂可拮抗吗啡作用。一旦停用吗啡或用纳络酮刺激,由于细胞内Ca2+浓度升高,产生戒断反应。本文主要介绍吗啡长期作用时不同分型的钙通道阻滞剂延缓吗啡成瘾发生的作用。
3.1VDCC VDCC是生物体内一大类钙通道,随着膜电位的改变而出现通道的开放、关闭和失活,调节细胞内Ca2+浓度,最终产生生物学效应。目前已知的VDCC有以下几型:L、N、P/Q、R、T 型(Tab 1)。
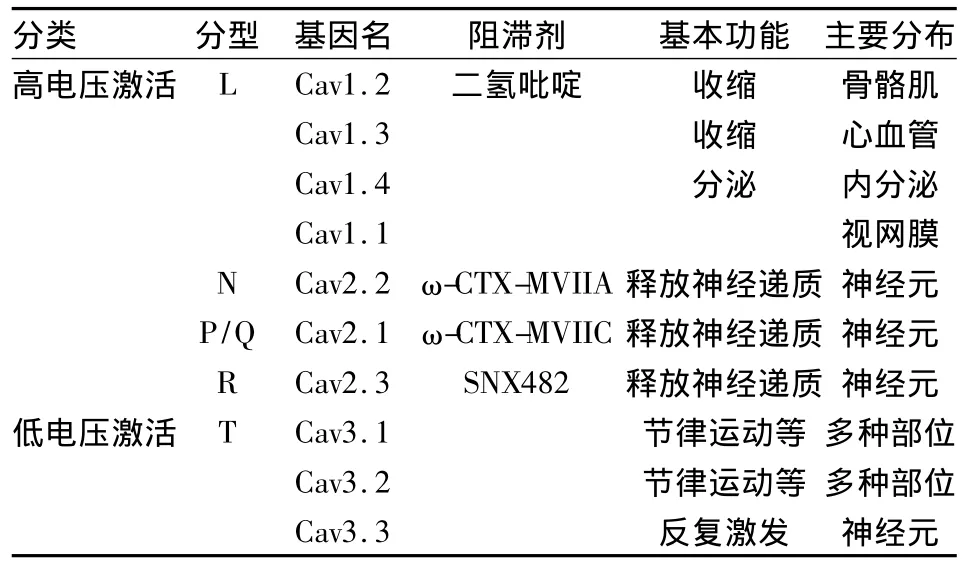
Tab 1 Different types,gene names,blockers,distributions and functions of the VDCC
大量研究证明VDCCs与阿片类药物成瘾的形成关系密切,VDCCs可通过多种途径参与阿片成瘾的形成。阿片类受体激动后,能够抑制VDCCs,使钙离子内流减弱。吗啡急性给药时,突触体的Ca2+浓度降低和Ca2+流入减少,而长期慢性给药形成依赖后突触囊泡内钙离子浓度水平明显提高,而且,依赖动物去除吗啡后戒断症状的维持时间和钙通道数目上调在时间上具有同步性,提示了两者之间的紧密联系。长期给予吗啡引起钙通道密度的增加,其浓度的增加也部分体现了动物对吗啡躯体依赖性的形成。
3.1.1L-型钙通道是高电压激活(HVA)通道(Cav1.1-Cav1.4),较强的去极化可开放,失活慢,广泛存在于各种兴奋细胞与非兴奋细胞膜上,介导各种细胞功能,对二氢吡啶类拮抗剂(如尼莫地平)高度敏感。研究资料表明,L-型钙通道拮抗剂能减轻吗啡依赖,翻转吗啡耐受。Verma等[2]报道L-型电压敏感性钙通道抑制剂尼莫地平和硝苯吡啶有效降低了大鼠对慢性吗啡摄入的耐受,免疫组化结果显示吗啡耐受模型中,钙通道的表达明显高于对照组,这也侧面证实了钙通道在依赖形成中发挥着重要作用。
另有研究证明,合用L-型钙通道阻滞剂尼莫地平可降低吗啡耐受的进展,推测尼莫地平抑制了吗啡诱导的L-型钙通道表达的上调从而降低了吗啡耐受[3]。而鞘内注射除尼莫地平外的其他L-型钙通道阻滞剂,不产生镇痛效应,尼莫地平也仅发挥短暂的镇痛效应[4],但当这些L-型钙通道阻滞剂(尼莫地平、硝苯地平、维拉帕米)与吗啡合用时,可显著增强吗啡的镇痛效应[3]。
3.1.2N-型钙通道也属于HVA型钙通道(Cav2.2),芋螺毒素ω-CTX-MVIIA,ω-CTX-GVIA可选择性阻断 N型钙电流,剂量依赖性的翻转异常疼痛[5]。慢性吗啡给药后N-型钙通道表达上调导致吗啡耐受,鞘内注射N-型钙通道阻滞剂氨氯地平(络活喜)(氨氯地平也可阻滞L-型钙通道)后可抑制吗啡耐受形成[6-7]。
本实验室前期研究证实N型钙通道拮抗剂ω-CTX MVIIA能够抑制吗啡诱导条件性位置偏爱(CPP)的建立,同时发现这种抑制作用与大鼠的运动活性和探索活动没有相关性,说明ω-CTX MVIIA对CPP的抑制并不是通过镇静作用和降低实验动物的运动活性实现的,而是通过抑制N型钙通道抑制了吗啡的奖赏效应及成瘾性。
Ziconotide(合成名为SNX-111)是ω-CTX-MVIIA的合成形式,为神经元特异性N型钙通道阻滞剂,有镇痛及神经元保护作用。鞘内注射可以阻断初级伤害感受器的传入神经递质的释放,同时阻止疼痛信号从脊髓向脑部传导,从而产生镇痛作用,且长期给药不会引起耐受性,优于鞘内注射吗啡。Wang等[8]发现ziconotide与吗啡合用短时间内有相加或协同作用,据此可降低吗啡用量,延缓耐受形成。长期鞘内注射ziconotide不产生耐药性,对吗啡也没有交叉耐药,但此药只有鞘内给药才能产生较好的疗效,并且存在其他缺点,如治疗窗口小,在剂量稍大时会出现眼球震颤、共济失调、恶心等副作用[9]。
模拟ω-芋螺毒素MVIIA(Ziconotide.SNX-111)的空间结构,Ge Meng等以4-氨基哌啶为母核,合成了一系列非肽类小分子化合物,并将其命名为ZC系列化合物,筛选出其中的ZC88具有特异性N型钙通道阻断作用且具有较好选择性。相关分析发现,ZC88可逆性地作用于N型钙通道的静息态和失活态,ZC88不仅对急性疼痛和慢性神经痛有明显镇痛作用,镇痛强度与其对N型钙通道的阻断能力有一定相关性,而且能够剂量依赖性地增强吗啡镇痛、对抗吗啡耐受和依赖[10]。
ω-芋螺毒素SO3是从线纹芋螺中获得的一种新型特异性N-型电压依赖型钙通道阻滞剂,鞘内重复给药不产生耐受,且与吗啡不发生交叉耐受,与吗啡合用时明显增加了其在福尔马林实验中的镇痛,在扭体实验中使剂量-反应曲线左移,吗啡重复给药后使得N-型钙通道表达上调导致吗啡耐受和敏化,但SO3在耐受小鼠中的镇痛作用不发生改变[11]。
虎纹镇痛肽-Ⅰ(Huwen analgesic peptide-Ⅰ,HWAP-Ⅰ)是从我国珍稀蜘蛛虎纹捕鸟蛛粗毒中分离纯化的一种多肽类神经毒素,与SNX-111具有相似的空间结构且具有N-型钙通道阻滞作用,均可剂量依赖性的抑制炎性内脏疼痛,虽然同等剂量下SNX-111镇痛效果略好,但较高剂量SNX-111会引起大鼠运动能力障碍,HWAP-I则无此副反应。吗啡镇痛效果优于二者但维持时间短[12]。
3.1.3P/Q-型钙通道属于中度高电压激活通道(Cav2.1),较L、N型通道失活慢,蜘蛛毒素可选择性抑制P型钙电流,目前尚未发现特异性Q型钙通道阻断剂,但它可被高浓度的ω-Aga-IVA大部分阻断,而1.5 mmol·L-1的ω-CTX-MVIIC则可完全阻断Q型钙电流。
3.1.4T-型钙通道属低电压激活(LVA)通道(Cav3.1、Cav3.2和 Cav3.3),快速激活,缓慢关闭。T-型钙通道阻滞剂咪拉地尔可增强吗啡的镇痛作用,且其对钙通道的阻滞降低了吗啡敏化从而抑制吗啡依赖和耐受的形成,并抑制了急性纳洛酮催促的戒断综合征[13]。阿片类药物能增加某些脑区突触体对Ca2+的摄取,增加钙内流的可能性,从而增加神经递质的释放,而T-型钙通道阻滞剂能抑制神经递质的释放以发挥镇痛作用。
3.2LGCC LGCC广泛分布于各种细胞,一般选择性较差,研究较多的是配体门控钙通道,此类通道是受体与通道本身就是一个复合体,当配体与受体结合后,通道开放,Ca2+内流。其中离子型谷氨酸受体:N-甲基-D-天冬氨酸受体(NMDAR)在吗啡成瘾中的作用日益引起人们的关注。研究已经证实[14]经典的非竞争性NMDA受体抑制剂MK-801不仅能够减轻吗啡诱发的躯体依赖症状,而且能够拮抗吗啡等精神活性物质的奖赏效应,但不能翻转已经形成的耐受。本实验室前期研究证明,作为NMDA受体的选择性拮抗剂,芋螺毒素中Conantokin家族中的Conantokin-G、Glu-Conantokin-G能够成功干预吗啡精神依赖。
3.3IP3受体三磷酸肌醇(IP3)是代谢型谷氨酸受体5(mGluR5)的下游信号分子,IP3与其受体(IP3R)结合后,促进内质网储存的Ca2+释放进入胞质,引起细胞内级联反应。IP3R介导的胞内钙释放依赖于一定浓度的胞质内Ca2+,呈现钟型反应(bell-shaped response):胞质内Ca2+浓度很低时,IP3R对IP3不敏感;Ca2+浓度上升到一定程度,IP3R最敏感;当胞质内Ca2+浓度进一步升高时,IP3R对IP3又不敏感。IP3R影响细胞代谢和功能,是形成吗啡耐受的关键因素[15]。
刘金变等[16]的研究证实吗啡作用7天后大鼠产生了耐受,此时吗啡已基本无镇痛效应。吗啡与代谢型谷氨酸受体拮抗剂(MPEP)合用延缓了镇痛效应的下降和耐受的产生。实验同时显示吗啡加MPEP组的IP3R-I的表达低于吗啡组,证明MPEP抑制吗啡耐受发展,机制可能与其抑制IP3R-I表达有关。
研究证明[17]吗啡激动阿片受体后通过PLC/IP3途径调节细胞内Ca2+浓度,脑室内注射IP3受体拮抗剂可剂量依赖性地抑制急性吗啡处理产生的镇痛效应。IP3受体的上调可能通过影响细胞内Ca2+浓度调节神经元可塑性变化,导致大鼠对吗啡的敏感性下降乃至消失,是形成吗啡耐受的细胞内信号机制之一。MPEP与吗啡合用组的IP3受体表达低于吗啡耐受组,单独鞘内应用MPEP对脊髓背角IP3受体的表达无影响,提示MPEP可能通过抑制吗啡耐受过程中IP3受体的上调抑制吗啡耐受发展。
此外有研究表明吗啡依赖小鼠给予东莨菪碱后,痛阈提高,跳跃次数减少,流式细胞术测量海马细胞内游离Ca2+浓度减少,证明东莨菪碱具有对抗吗啡依赖性的作用,其作用机制可能与降低脑内游离Ca2+水平有关。在吗啡依赖和戒断综合征期间,内源性乙酰胆碱释放增多,M型胆碱能受体通过激活磷脂酶C(PLC)分解膜磷脂磷酯酰肌醇二磷(PIP2)产生IP3,使细胞膜对Ca2+的通透性增加,细胞内Ca2+浓度升高[18]。升高的细胞内Ca2+浓度又可引起神经递质释放量增加,感觉冲动传入增加,导致痛阈降低、躯体依赖和戒断症状。而东莨菪碱是M型胆碱能受体拮抗剂,可通过阻断IP3-Ca2+途径,降低细胞内Ca2+浓度,从而提高痛阈减轻躯体依赖和戒断症状。有文献报道阿片在成瘾过程中细胞内Ca2+的浓度进行性增高,因此,一般认为细胞内Ca2+浓度水平可以成为吗啡成瘾程度的一个客观指标,而东莨菪碱可能在中枢胆碱能神经元的Ca2+内流及递质释放两者相互关联的中枢环节中起着重要的调节作用[19]。
3.4雷诺定受体目前对雷诺定受体与吗啡依赖作用的研究未见报道,但Galeotti等[20]研究发现雷诺定受体与其生理性配体环腺苷二磷酸核糖(cADPR)结合后,释放钙池内Ca2+进入胞质;抑制雷诺定受体后细胞质内Ca2+的浓度可明显降低,并可明显延缓疼痛的产生,提示雷诺定受体在镇痛中发挥作用。
3.5其他除上述钙通道阻滞剂外,我国中药宝库中许多药物也可以通过抑制胞外钙内流,减轻钙超载缓解吗啡戒断症状。
三七总皂苷(panax notoginsebg,PNS)是中药三七的提取物,已有资料证实[21]PNS可通过钙通道阻断作用减轻脑和脊髓损伤,应用PNS后亦可抑制吗啡依赖和纳洛酮催促时出现的戒断症状,分析原因可能是:抑制了戒断时胞外钙异常内流和钙超载,减少Ca2+和钙调蛋白(CaM)结合,抑制Ca2+-CaM-CaMK通路亢进和表达增高,从而有效的抑制了大鼠海马组织纳洛酮催促戒断引起的钙调蛋白激酶Ⅱ(CaMKⅡ)活性和CaMKⅡα亚基蛋白异常升高,从而抑制了吗啡依赖的发生[22]。对三七的主要成分三七总皂苷Rg1的研究也发现[23]:Rg1可能通过抑制[Ca2+]i的升高,使Ca2+/CaM复合物减少,抑制CaMKⅡβ mRNA和蛋白的表达,阻断吗啡成瘾及戒断的产生。
4 结语
钙通道参与吗啡镇痛和成瘾作用。由于钙通道阻滞剂具有较好的镇痛效果,且不具成瘾性,又可抑制成瘾的形成和减轻戒断症状,钙通道已成为一个重要的药物研究靶标。对钙通道及其阻滞剂的深入研究,将有助于人们对成瘾机制的认识和吗啡成瘾治疗药物的研发。
[1]Bemardi P.Mitochondrial transport of cations:channels,exchangers,and permeability transition[J].Physiol Rev,1999,79(4):1127-55.
[2]Verma D,Gupta Y K,Parashar A,Ray S B.Differential expression of L-and N-type voltage-sensitive calcium channels in the spinal cord of morphine+nimodipine treated rats[J].Brain Res,2009,1249:128-34.
[3]Ray S B,Mishra P,Verma D,et al.Nimodipine is more effective than nifedipine in attenuating morphine tolerance on chronic co-administration in the rat tail-flick test[J].Indian J Exper Biol,2008,46:219-28.
[4]Gupta H,Verma D,Ahuja R K,et al.Intrathecal co-administration of morphine and nimodipine produces higher antinociceptive effect by synergistic interaction as evident by injecting different doses of each drug in rats.[J].Eur J Pharmacol,2007,561(1-3):46-53.
[5]Matthews E A,Dickenson A H.Effects of spinally delivered N-and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy[J].Pain,2001,92(1-2):235-46.
[6]Murakami M,Nkagawasai O,Fujii S,et al.Antinociceptive action of amlodipine blocking N-type Ca2+channels at the primary afferent neurons in mice[J].Eur J Pharmacol,2001,419(2-3):175-81.
[7]Dogrul A,Bilsky E J,Ossipov M H,et al.Spinal L-type calcium channel blockade abolishes opioid-induced sensory hypersensitivity and antinociceptive tolerance[J].Anesth Analg,2005,101(6):1730-5.
[8]Wang Y X,Gao D,Pettus M,et al.Interactions of intrathecally administered ziconotide,a selective blocker of neuronal N-type voltage-sensitive calcium channels,with morphine on nociception in rats[J].Pain,2000,84(2-3):271-81.
[9]Wermeling D P.Ziconotide,an intrathecally administered N-Type calcium channel antagonist for the treatment of chronic pain[J].Pharmacotherapy,2005,25(8):1084-94.
[10]Meng G,Wu N,Zhang C,et al.Analgesic activity of ZC88,a novel N-type voltage-dependent calcium channel blocker,and its modulation of morphine analgesia,tolerance and dependence[J].Eur J Pharmacol,2008,586(1-3):130-8.
[11]吉小莉,颜玲娣,周培岚等.芋螺毒素ω-SO3单次及连续给药对福尔马林致大鼠炎性疼痛的阵痛作用[J].中国药理学通报,2010,26(4):476-82.
[11]Ji X L,Yan L D,Zhou P L,et al.Antinociception of omega-conotoxin ω-SO3 in rat formalin test after acute or chronic intrathecal administration[J].Chin Pharmacol Bull,2010,26(4):476-82.
[12]陈嘉勤,陈威华,邓梅春,等.一种新型N-型电压敏感性钙通道拮抗剂虎纹蜘蛛毒素-Ⅰ对大鼠内脏痛模型阵痛作用的研究[J].第一军医大学学报,2005,25(1):10-4.
[12]Chen J Q,Chen W H,Deng M C,et al.Analgesic effect of huwentoxin-I,a new N-type voltage-sensitive calcium channel blocker,on acute visceral pain in rats[J].J First Mil Med Univ,2005,25(1):10-4.
[13]Dogrul A,Zagl U,Tulunay F C.The role of T-type calcium channels in morphine analgesia,development of antinociceptive tolerance and dependence to morphine,and morphine abstinence syndrome[J].Life Sci,2002,71(6):725-34.
[14]Shu H H,Hayashida M,Huang W Q,et al.The comparison of effects of processed aconiti tuber,U50488H and MK-801 on the antinociceptive tolerance to morphine[J].J Ethnopharmacol,2008,117(1):158-65.
[15]Gabra B H,Smith F L,Navarro A,et al.mGluR5 antagonists that block calcium mobilizationin vitroalso revere(S)-3,5-DHPG-induced hyperalgesia and morphine antinociceptive tolerancein vivo[J].Brain Res,2008,1187:58-66.
[16]刘金变,江 伟.MPEP对吗啡耐受大鼠脊髓Ⅰ型IP3受体表达的影响[J].中国药物依赖性杂志,2009,18(1):24-7.
[16]Liu J B,Jiang W.Effect of MPEP on the expression of IP3R-Ⅰ in the spinal cord of morphine tolerant rats[J].Chin J Drug Depend,2009,18(1):24-7.
[17]Aoki T,Narita M,Ohnishi O,et al.Disruption of the type 1 inositol 1,4,5-trisphosphate receptor gene suppresses the morphine-induced antinociception in the mouse[J].Neurosci Lett,2003,350(2):69-72.
[18]Volkow N D,Fowler J S,Wang G J.The addicted human brain viewed in the light of imaging studies:brain circuits and treatment strategies[J].Neuropharmacology,2004,47:3-13.
[19]王黎光,郭新华,刘凌云,等.东莨菪碱抗吗啡依赖作用与海马细胞内钙的关系[J].中国应用生理学杂志,2006,22(3):307-9.
[19]Wang L G,Guo X H,Liu L Y,et al.Effect of Scopolamin on Morphine-dependence in mice and the relationship between the effect and Hippocampus intracellular calcium[J].Chin J Appl Physiol,2006,22(3):307-9.
[20]Galeotti N,Bartolini A,Ghelardini C.Ryanodine receptors are involved in muscarinic antinociception in mice[J].Behav Brain Res,2005,164(2):165-71.
[21]郭庆民,刘景生.阿片类药物对 NG108-15细胞Ca2+/钙调蛋白依赖的蛋白激酶Ⅱ信息通路的作用[J].药学学报,2001,36(9):652-6.
[21]Guo Q M,Liu J S.Effects of opioids on Ca2+/calmodulin dependent protein kinase signal pathway in NG108-15 cells[J].Acta Pharm Sin,2001,36(9):652-6.
[22]牛增强,闫玉仙,马春玲,等.三七总皂甙对吗啡戒断大鼠海马组织CaMKII活性及CaMKIIα亚基蛋白表达的影响[J].中国药物依赖性杂志,2008,17(2):102-5.
[22]Niu Z Q,Yan Y X,Ma C L,et al.Effects of panax notoginseng on CaMKII activity and CaMKIIα protein expression in hippocampus of morphine withdrawal rats[J].China J Drug Depend,2008,17(2):102-5.
[23]闫玉仙,宋月英,王小平,等.人参皂苷Rg1对慢性吗啡作用及纳洛酮催促戒断SK-N-SH细胞的影响及机制研究[J].中国药理学通报,2010,26(8):1074-8.
[23]Yan Y X,Song Y Y,Wang X P,et al.Effects and mechanism of ginsenoside-Rg1 on SK-N-SH cell treated with chronic morphine and naloxone-precipitated withdrawal[J].Chin Pharmacol Bull,2010,26(8):1074-8.
猜你喜欢
介入放射学杂志(2022年10期)2022-11-02
科技创新与应用(2021年7期)2021-02-04
医药前沿(2020年23期)2020-12-03
智富时代(2019年6期)2019-07-24
智富时代(2019年6期)2019-07-24
中成药(2018年10期)2018-10-26
中国药理学与毒理学杂志(2017年4期)2017-06-01
中西医结合心血管病杂志(电子版)(2015年35期)2015-01-21
文史博览·文史(2014年11期)2015-01-14
中国药理学通报(2013年11期)2013-12-08
