
童年期发病的Leber遗传性视神经病变临床研究
2016-04-08 06:52宫晓红韦企平周剑夏燕婷孙艳红陈亚娟
中国中医眼科杂志 2016年6期
宫晓红,韦企平,周剑,夏燕婷,廖 良,孙艳红,陈亚娟
童年期发病的Leber遗传性视神经病变临床研究
宫晓红,韦企平,周剑,夏燕婷,廖 良,孙艳红,陈亚娟
目的探讨童年期发病的Leber遗传性视神经病变(LHON)患者线粒体DNA(mtDNA)突变的主要类型、临床特征及预后。方法2000年6月—2015年6月在我院眼科就诊且经实验室检查已确诊为LHON的患者279例,对其中≤16岁的童年期发病LHON患者共计161例进行临床资料归纳分析。结果161例童年期发病LHON中11778位点突变者144例(89.4%),其中3例合并14502位点突变,1例合并11696位点突变,1例合并14693位点突变。14484位点突变者11例(6.8%),继发位点突变6例(3.7%),其中3316位点突变2例,14693位点突变2例,11696位点突变1例,3497位点突变1例。未检测到3460等其他常见原发突变位点。童年期发病LHON典型临床表现为:双眼先后或同时中心视力急性或缓慢下降,多不伴眼球疼痛,视野检查以中心或旁中心暗点为主。随访视力恢复至≥0.3的儿童LHON患者中,11778位点突变者104只眼中有13只眼,占12.5%,14484位点突变者6只眼中有4只眼,占66.7%。童年期发病的LHON患儿中部分低年龄段儿童初诊视力及最终视力恢复比大龄儿童好。结论童年期发病LHON患儿其主要突变位点发生率及临床特征与成人LHON患者相似,其中部分低年龄段儿童视力恢复较好。童年期发病LHON患者应与其他遗传性视神经疾病及儿童视神经炎相鉴别。
童年期;Leber病;临床特征;鉴别诊断
Leber遗传性视神经病变(Leber hereditary optic neuropathy,LHON)是临床上少见的双眼中心视力先后无痛性、亚急性下降的视神经病变。我科曾有临床相关报道[1],占据本病大多数的11778基因位点突变的LHON患者(167例,84%)的发病年龄范围在2~45岁之间,平均发病年龄(16.9±6.3)岁,其中发病年龄≤15岁LHON患者有84例(84/167,50%)。国际《儿童权利公约》将儿童定义为18岁以下,但参照医学统计惯例,我们将儿童年龄减小到16岁,加上近4年收集的病例,现将共计161例童年期发病的LHON患者的临床资料归纳总结如下。
1 对象和方法
1.1 研究对象
2000年6月—2015年6月在我院眼科门诊及住院治疗的≤16岁的LHON患者161例(322只眼),均为童年期发病,且线粒体DNA(mtDNA)基因检测结果阳性。
临床检查:包括最佳矫正视力(Snellen视力),眼前节、眼底、眼压及色觉检查,视野(Octopus101视野计(视力≥0.1,可配合者采用tG2程序检查和统计,能识别LVC大光标、但不能配合tG2程序者采用LVC程序检查和统计),光学相干断层扫描(OCT)(蔡司公司),图形视觉诱发电位(P-VEP)或闪光视觉诱发电位(F-VEP)(德国罗兰双通道电生理仪,视力≥0.1采用P-VEP检查,视力<0.1采用F-VEP检查),荧光素眼底血管造影(FFA)等。常规眼科检查由指定医师完成,并询问家族史,绘制家系图。
治疗方法:门诊以口服中药和/或针灸治疗为主;住院患儿增加静脉滴注改善循环药物及肌注营养神经药物。部分患儿加用艾地苯醌(Idebenone),每次口服30~60 mg(1~2片)。总疗程2~6个月,平均3个月。
1.2 数据分析
对纳入患者的一般情况、mtDNA阳性位点构成、常规及仪器检查的初诊和随访结果进行统计。Snellen视力转换为最小分辨角的对数视力(Log-Mar),即Snellen视力1.0=LogMar视力0,数指=2.0,手动=3.0,光感=4.0,无光感=5.0。采用SPSS 20.0软件对数据进行统计学处理,定量资料符合正态分布者以均值±标准差(x±s)表示,否则以中位数及四分位数[M(Q1,Q3)]表示,初诊及末次随访检查结果比较采用配对样本Wilcoxon带符号秩检验,以P<0.05为差异有统计学意义。
2 结果
2.1 初诊情况
基因位点检测结果:161例童年期发病的LHON患儿中11778位点突变者144例,占89.4%,其中3例合并14502位点突变,1例合并11696位点突变,1例合并14693位点突变;14484位点突变者11例,占6.8%;继发位点突变6例,占3.7%,其中3316位点突变2例,14693位点突变2例,11696位点突变1例,3497位点突变1例;未检测到3460位点突变。
一般情况:161例患儿中,男137例,女24例,男女之比为5.7∶1。其中有母系家族史的86例(53.4%)。发病年龄2~16岁,平均年龄(12.46±3.63)岁。0~6岁者18例,7~12岁者38例,>12岁者105例。病程3 d~197个月,平均病程90(27,365)d。双眼先后发病89例,自觉同时发病57例,15例发病情况不明。视力急性或亚急性下降93例,逐渐下降47例,另有21例均为偶然发现(如入托儿所、入学及娱乐中被发现低视力)。发病时仅有11例患儿自述有眼痛或眼胀不适。
视力和色觉:除23例无视力记录外,138例276只眼中,眼前数指或手动者22只眼,指数~0.1者95只眼,0.1~0.3者129只眼,0.4~0.6者21只眼,>0.6的9只眼。色觉检查67例中,红绿色盲22例,色弱37例,全色盲1例,正常7例。
眼底:在322只眼中视盘色泽正常37只眼,视盘充血状伴有盘周中小血管扩张及神经纤维层发灰模糊59只眼(其中1例右眼视盘下方有火焰状出血,见图1)。视盘变淡或苍白以颞侧为主226只眼。检查黄斑区正常67只眼,中心凹光反射不清或消失209只眼,色素不均或紊乱44只眼,锡箔样反光2只眼。
特殊检查:视野检查71例142只眼(其中44只眼视力<0.1的未能准确评价视野),实际检查结果98眼次。包括中心暗点46只眼,旁中心暗点35眼,盲中心暗点17只眼,不同象限或神经纤维束样缺损15眼,向心性缩小14只眼,仅生理盲点扩大3只眼,局限性或弥漫性压陷各2只眼,不规则缺损10只眼。对病程小于3个月,视盘仍红润的24例48只眼行FFA检查,其中23只眼显示视盘和其周围血管明显扩张,但无荧光渗漏;9只眼的视盘荧光充盈饱满,晚期荧光素潴留仍无渗漏;2只眼在扩张的血管壁有荧光素壁染,8只眼视盘荧光减弱,6只眼未发现异常。电生理P-VEP或F-VEP(视力低于0.1)检查44例88只眼,结果P100波潜伏期延迟40只眼,正常2只眼;振幅降低38只眼,正常4只眼,无波形4只眼。初诊15例采用OCT进行视网膜神经纤维层(retinal nerve fiber layer,RNFL)厚度的扫描,表现为患儿RNFL增厚,右眼(125.13±30.85)μm,左眼(117.53±30.11)μm。
伴随的眼部或全身情况:发病伴随光幻觉4例,耳鸣6例,头痛2例,癫痫病史2例,上颌窦炎2例,眼痛胀1例,垂体微腺瘤1例,右手及前臂先天缺失1例,内眦宽、类似鸟脸畸形伴外斜并伴佝偻病、O型腿1例,双眼先天性白内障、新生儿脑炎、病毒性心肌炎(已愈)、窦性心律不齐及预激综合征各1例。另有5例感冒后发病,2例劳累后发病及1例情绪紧张后发病,但均不能确认是否为LHON的诱发因素。
2.2 随访结果
视力:截止到2015年12月,平均随访184(139,328)d。随访资料完整的住院患者55例(110只眼),67只眼视力提高1行或无改善;17只眼视力提高2至3行,26只眼视力提高大于3行。末次随访双眼视力与初诊视力比较均有统计学差异(P<0.001)(表1)。

表1 55例LHON患儿治疗前后视力比较[M(Q1,Q3)](LogMar视力)
OCT:截止到2015年12月,平均随访600(77,931)d。对照20例40只眼初诊和随访RNFL厚度扫描提示,患者双眼RNFL均有明显薄变(表2)。
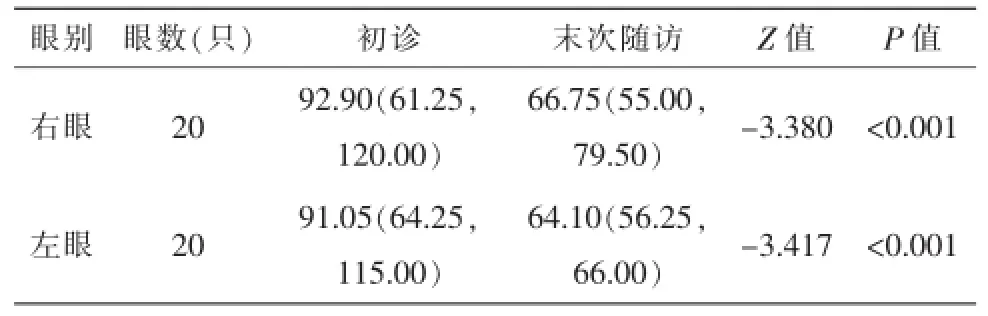
表2 20例LHON患儿的OCT视盘RNFL平均厚度随访结果[M(Q1,Q3),μm]
P-VEP:截止到2015年12月,平均随访600(207,902)d。14例28只眼检测结果提示,患者P100波潜伏期及振幅较初诊无明显变化(P>0.05)(表3)。F-VEP结果因变异度过大未纳入统计。

表3 14例LHON患儿P-VEP P100波潜伏期及振幅随访结果[M(Q1,Q3)]
视野检查:截止到2015年12月,平均随访779(490,904)d。对19例(35只眼)视野检查数据随访,其中11例(21只眼)随访前后视力均≥0.1,采用tG2模式检查和统计;8例(14只眼)随访前后视力均小于0.1,采用LVC模式检查和统计。数据统计结果提示上述35只眼末次随访视野与初始视野均无明显差异(表4)。另外,随访期间LVC程序转tG2程序检查者5只眼、由tG2模式转LVC模式2只眼,因不同视野程序参考值存在差异,故此7眼未行数据统计。
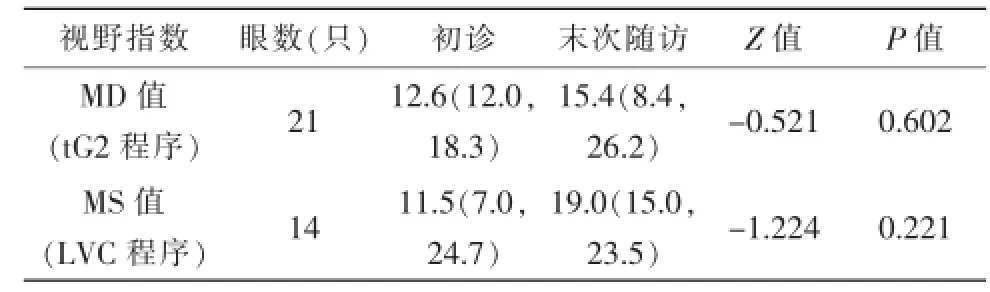
表4 19例LHON患儿的视野指数随访结果[M(Q1,Q3),dB]
3 讨论
1988年,Wallace等[2]首先证实Leber病是由mtDNA突变造成的。目前,国内外报告涉及LHON的mtDNA突变基因位点已有40多个,其中11778,3460,14484仍是占主流的3个原发性突变[3]。其他突变位点如15257、14693等则称为继发性突变。本研究中144例11778位点突变的LHON患儿,占mtDNA突变患儿的89.4%,11例14484位点突变的LHON患儿,占mtDNA突变儿童患者的6.8%,继发位点突变6例,占3.7%,未发现其他位点突变,提示童年期发病的LHON患者中仍以11778位点突变占绝大多数。
Oguchi[4]曾报告视力恢复≥0.3的LHON患者11778位点突变中仅占7%,14484位点突变中占60%。本组中视力恢复≥0.3的患儿中11778位点突变占12.5%,14484位点突变占66.7%,研究结果还发现,55例(110只眼)LHON患儿中年龄≤12岁(21例)的初诊及治疗后视力均较>12岁(34例)者为好,是否提示发病早的儿童视力恢复率较高仍待更多病例证实。对于童年期发病的LHON,视力预后相对较好,视野亦有以中心暗点开窗或缩小为特征的改善已有文献报告[5-6],笔者将以前瞻性对照研究做进一步证实。总结前述161例童年期发病的LHON患儿临床特征:仍以男性为主;双眼先后或同时视力急剧或亚急性下降,也可视力缓慢下降,98.4%的患者无眼球疼痛感;视力<0.1者居多(77.3%),未见失明者;多数患者伴有红绿色觉障碍;视野有中心、旁中心、盲中心暗点;眼底检查早期正常或视盘充血、盘周微血管迂曲扩张,FFA病变区无染料渗漏,晚期视神经萎缩;视觉诱发电位检查P100波潜伏期延迟,振幅下降。值得注意的是部分(32.7%)童年期发病LHON患儿因病初双眼有类似视乳头炎表现,曾先后在数家医院按视神经炎用激素治疗为主,最终在我院行基因检测确诊。提示我们对于临床诊断为双眼视乳头炎的儿童病例应注意与LHON鉴别;对于临床特征疑似LHON的儿童,即便年龄很小也应常规行mtDNA检测;对于临床特征不典型但病因不明者也应行mtDNA检测;此外还应与常染色体显性视神经萎缩(autosomal dominant optic atrophy,ADOA)鉴别,我科近年已关注本病[7],对怀疑遗传性视神经病变的9例儿童经mtDNA检测已排除LHON,进一步行OPA1基因检测证实是ADOA。后者发病多在10岁前,4~8岁常见。临床特点是双眼视力无痛性缓慢进展性下降,可降至0.1~0.3,偶见仅剩数指者或仍维持在1.0的。眼底双眼颞侧视盘苍白,色觉障碍有特征性的蓝-黄色觉缺损及视野有中心或盲中心盲点等[8]可与LHON鉴别。
我们的病例均采用中药结合针刺治疗为主,部分病例加用艾地苯醌口服。虽然总的疗效高于国外报告,但因未设分组对照研究,中医的确切疗效尚待证实。国外近年采用基因治疗Leber先天性黑朦取得部分疗效[9-10]。国内Xing Wan等在前期动物实验基础上[11],又在临床上对9位11778位点突变的LHON患者单眼注射重组腺相关病毒2介导的ND4基因制剂,随诊9个月,结果:6位患者视力至少提高0.3 LogMAR,视野扩大,OCT检查神经纤维层厚度没有明显变化。在9个月的观察时间里,9位患者没有发现任何全身和局部的不良反应。该研究认为采用基因治疗LHON是可行的[12]。国际上相关基础和临床基因干预治疗部分遗传性眼病的研究仍在安全性和有效性及机理诸方面的深入探讨中。
[1]韦企平,孙艳红,周翔天,等.Leber遗传性视神经病变临床研究[J].中华眼科杂志,2012,48(12):1065-1068.
[2]Wallace DC,Singh G,Lott MT,et al.Mitochondrial DNA muta-tion associated with Leber’s hereditaty optic neuropathy[J].Science,1988,242(4884):1427-1430.
[3]Mojon DS,Herbert J,Sadiq SA,et al.Leber’s hereditary optic neuropathy mitochondrial DNA mutations at neucleotides 11778 and 3460 in multiple sclerosis[J].Ophthalomologica,1999,213(3):171. [4]Oguchi Y.Past,present,and future in Leber’s hereditary optic neuropathy[J].Nippon Ganka Gakkai Zasshi.2001,105(12):809-827. [5]Barboni P,Savini G,Valentino ML,et al.Leber’s Hereditary Optic Neuropathy with Childhood Onset[J].IOVS,2006,47(12):5303-5309.
[6]Acaroglu G,Kansu T,Dogulu C.Visual recovery patterns in children with Leber’s hereditary optic neuropathy[J].International Ophthalmology,2001,24(6):349-355.
[7]韦企平(专题述评).应重视儿童遗传性视神经萎缩的临床研究[J].中国眼耳鼻喉科杂志,2013,13(4):211-213.
[8]Maresca A,La Morgin C,Caporali L,et al.The optic nerve.A“mitowindow”on mitochondrial neurodegeneration[J].Mol Cell Neurosci. 2013,55.62-76.
[9]Maguire AM,Simonelli F,Pierce EA,et al.Safety and efficacy of gene transfer for Leber’s congenital amaurosis[J].N.Engl.J.Med. 2008,358(21):2240-2248.
[10]Bainbridge JW,Smith AJ,Barker SS,et al.Effect of gene therapy on visual function in Leber’s congenital amaurosis[J].N.Engl.J.Med. 2008,358(2):2231-2239.
[11]Shi H,Gao J,Pei H,Liu R,et al.AAV-mediated gene delivery of the human ND4 complex I subunit in rabbit eyes[J].Clin Experiment Ophthalmol,2012,40(9):888-894.
[12]Xing Wan,Han Pei,Min-jian Zhao,et al.Efficacy and Safety of rAAV2-ND4 Treatment for Leber’s Hereditary Optic Neuropathy [J].Scientific Reports,2016,6:1-10.
A clinical study of childhood-onset Leber’s hereditary optic ueuropathy
GONG Xiaohong,WEI Qiping,ZHOU Jian,et al.
Dongfang Hospital,Beijing University of Chinese Medicine,Beijing 100078,China
OBJECTIVE To investigate the main mitochondrial DNA(mtDNA)mutation types,clinical characteristics and the prognosis of Leber’s Hereditary Optic Neuropathy(LHON)onset in childhood.METHODS A total of 161 cases with childhood-onset LHON aged 16 years or younger were selected from 279 cases and then analyzed,all of whom were diagnosed with laboratory proof.RESULTS Among the 161 cases,G11778A primary mutation occurred in 144 cases(89.4%),while 3 of them was complicated with 14502 mt-DNA mutations,1 of them with 11696 mt-DNA mutation in addition,and 1 of them was accompanied with the 14693 mt-DNA mutation.And G14484A primary mutation occurred in 11 cases(6.8%).Besides,6 cases(3.7%)were identified with secondary mutations,among which 2 cases were of 3316 mt-DNA mutation,2 cases were of 14693 mt-DNA mutation,1 case was of 11696 mt-DNA mutation and 1 case was of 3497 mt-DNA mutation.However,some classic primary mutations such as 3460 mutation were not found.Childhood-onset LHON typically manifested symptoms and signs as:acute or subacute painlessly central vision loss in both eyes at the same time or one eye after another;a central or para-central scotoma in visual field test.In this study,13 eyes(12.5%)of 104 with G11778A mutation and 4 eyes(66.7%)of 6 with G14484A mutation restored visual acuity to 0.3 or better.In addition,younger cases showed better restorative visual acuity than elder cases both in initial and follow-up test.CONCLUSIONS The main mt-DNA mutation types and clinical characteristics of childhood-onset LHON patients were similar with adult-onset patients. Onset in younger age often implied lower damage and better prognosis.The childhood-onset LHON should be differentiated from other hereditary optic neuropathy and children optic neuritis.
childhood;Leber’s hereditary optic neuropathy;clinical characteristics;differential diagnosis
R774.6
A
1002-4379(2016)06-0355-04
10.13444/j.cnki.zgzyykzz.2016.06.002
北京中医药大学东方医院眼科,北京100078
韦企平,E-mail:wei_dfyy@163.com
猜你喜欢
山东医药(2021年14期)2021-05-27
临床眼科杂志(2021年2期)2021-05-26
科学(2020年3期)2020-11-26
中华民居(2020年3期)2020-07-24
中医眼耳鼻喉杂志(2019年3期)2019-04-13
中医眼耳鼻喉杂志(2018年1期)2018-04-10
中国中医眼科杂志(2015年1期)2015-12-28
中国中医眼科杂志(2015年1期)2015-12-28
科学家(2015年2期)2015-04-09
读者(2014年18期)2014-05-14
