
N2H4分子取代基效应的量子化学研究
2017-05-15 10:56郭雅琼高小童贾亚兰
四川师范大学学报(自然科学版) 2017年1期
郭雅琼, 高小童, 贾亚兰, 张 懿, 毛 双*
(1. 四川师范大学 化学与材料学院, 四川 成都 610066; 2. 武警警官学院, 四川 成都 610213)
N2H4分子取代基效应的量子化学研究
郭雅琼1,2, 高小童1, 贾亚兰1, 张 懿1, 毛 双1*
(1. 四川师范大学 化学与材料学院, 四川 成都 610066; 2. 武警警官学院, 四川 成都 610213)
计算了甲基(—CH3)和羟基(—OH)对N2H4的取代基效应.引入甲基后,N2H4的N—N键长变长,电荷密度变小.羟基的引入,使得N—N键长变短,其中1,1,2,2-四羟基N2H4的N—N键长显著变化.通过NBO计算,N—N键的键级随着取代基个数的增加逐渐减小,超共轭作用在决定构型相对稳定性方面起了重要作用.引入取代基后,N原子的孤对电子与N—C(N—O)键之间发生相互作用,使得整个分子的超共轭作用增强.随着取代基数目的增多,分子的总能量和生成热均降低,取代基数目与分子相对稳定性之间具有较好的相关性.
量子化学; 取代基效应; 氮氢化合物; 异构体; 相对稳定性
20世纪90年代初,高能量密度材料(HEDM)正式进入发展阶段.随着时间的推移以及高科技技术的不断进步,因为可用作制造炸药、推进剂或火工品的材料,高能量密度材料几乎被所有武器系统所使用,在现代科技工业中发挥了极为重要的作用[1-3].由于氮氢化合物(NnHm)中存在N—N基团,氮原子的孤对电子产生相互排斥作用,使得氮氢化合物不稳定且具有高的生成焓,在自然界中比较罕见,大多数都以反应中间体或裂解产物形式存在[4-5].但是因为此类化合物在含能材料方面的重要性,从20世纪50年代,人们开始着手对氮氢化合物的理论和实验研究[6-7].目前,关于其研究主要是以理论为主.W. B. David[8-9]对N4H4和N4H6系列分子的几何构型构象变化以及质子亲和势等方面进行了理论研究.高氮化合物作为新型的含能材料,Los Alamos国家实验室的Hiskey研究小组[10]对其合成以及应用也曾进行了大量的研究.文献也报道过运用G3B3方法探索N4H4、N3H3的几何构型、生成热、稳定性和互变异构现象[11-12].总之,对氮氢化合物的研究,随着体系的增大,数量会越来越多.肼(N2H4,hydrazine)在氮氢化合物中是比较简单的一种物质,它与它的衍生物构成了一系列很重要的含能化合物,既可用作钢铁的防腐剂材料,又能用作卫星和火箭推动器的燃料[13-15].借助已有的对氮氢化合物的理论研究,发现在其上引入其他基团后,分子的部分性质会发生变化.本文主要探索N2H4的H分别被甲基、羟基取代后的衍生物相对于N2H4的几何构型及能量的变化规律.
1 计算方法
应用密度泛函理论的B3LYP方法在6-311++g**基组下对N2H4可能存在的甲基和羟基取代异构体进行了几何优化和振动分析.结果表明,计算所得到的构型均为势能面上的稳定点.同时用AIM 2000程序包[16]对化合物的键临界点电荷密度进行了计算,明确了化学键的性质.在相同基组水平上采用自然键轨道(NBO)[17]分析方法对几何构型进行了分析,揭示了超共轭作用对于取代物构型稳定性的影响.最后采用G3MP2方法对分子能量进行校正,并计算了各异构体在298 K时的生成热,所有计算都采用Gaussian 09程序[18]完成.
2 结果与讨论
2.1 几何性质 图1列出了N2H4及甲基异构体,其中(a)为键长(nm),(b)为键临界点处的电荷密度ρ(a.u.),(c)为2ρ(a.u.),(d)为键级.在这里选择下列构型:1-甲基N2H4(B),1,1-二甲基N2H4(C),1,1,2-三甲基N2H4(E)和1,1,2,2-四甲基N2H4(F),来讨论取代基的加入对N2H4分子的影响.在这4个构型中H原子逐步被甲基(—CH3)所取代.
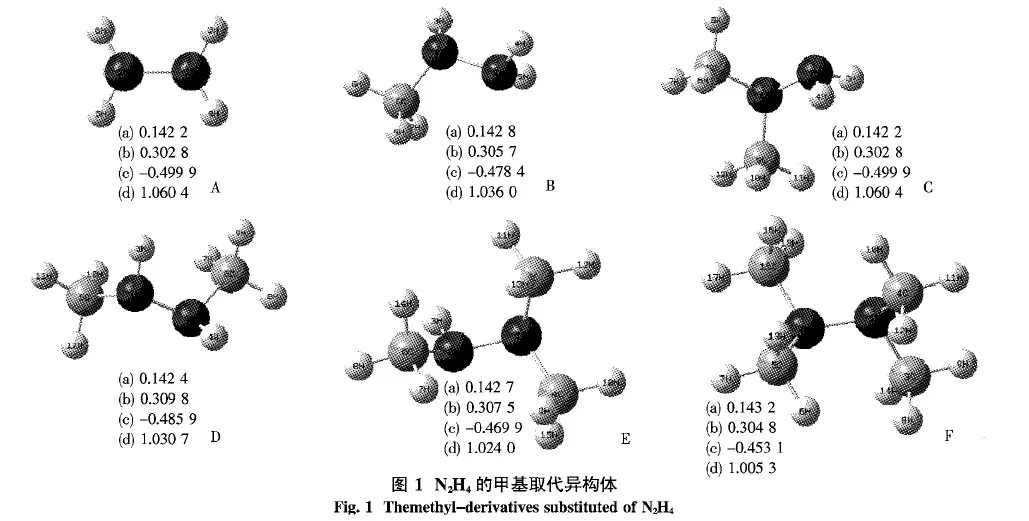
表 1 化合物异构体的主要二阶稳定化能分析参数

化合物BD-LPE(2)/(kJ/mol)A(N3)—(N2—H6)5.98(N2)—(N3—H5)6.02B(N1)—(C6—H7)7.18(N1)—(N2—H5)5.59(N2)—(N1—H3)4.86C(N1)—(C9—H10)8.46(N1)—(N2—H4)7.40(N2)—(N1—C9)6.12D(N1)—(C5—H10)7.37(N1)—(N2—C6)6.34(N2)—(C6—H9)7.23E(N1)—(C5—H13)8.46(N1)—(C4—H9)8.13(N2)—(N1—C5)7.43(N1)—(C3—H8)8.31F(N2)—(C15—H16)8.30(N2)—(C5—H13)8.28
图1中分别列出了N2H4及4种甲基(—CH3)取代物的键长、AIM和键级分析.从键长数据可以看出,随着甲基(—CH3)数目的增多,N—N的键长呈逐渐增大的趋势,甲基(—CH3)取代异构体的N—N键长均大于N2H4(A)的N—N键长,同时经AIM分析发现其所有构型的2ρ<0,表明所有的化学键均为共价键.通过NBO计算了键级,发现随着取代基个数的增加,2个N原子间的键级在逐渐减小.
为了进一步了解超共轭作用的实质,进行了二阶稳定化能的分析.由表1可知,N2H4分子中的超共轭作用主要体现在N原子孤对电子与相邻的N—H键、N—N键之间.而当H原子被甲基逐步取代后,分子中就产生了N原子孤对电子与相邻的N—C键、N—H键、N—N键之间的相互作用,随着甲基数目的增多,二阶稳定化能呈增大的趋势.
2.2 能量性质 表2中列出了计算所得的各异构体的能量,其中ENL为离域能,EL为LUMO轨道能,EH为HOMO轨道能,△E:=EL-EH,E(L)为超共轭缺失的总能,△fH0为生成热.从表2的数据可以看出,随着取代基数目的增多,分子总能量逐渐降低,每增加一个甲基(—CH3),分子总能量就相应降低39.3 a.u..N2H4(A)的二取代物有2种(C,D),其中C的分子能量较低,是稳定构型.分子的总能量与其相对稳定性有关,而生成热则是衡量高能材料爆炸性能的重要参数.因此采用G3MP2方法,在Pθ、298 K下计算了各个异构体的生成热.由计算数据可以看出,这些异构体的生成热均为正,它们都是吸热材料,均具有了含能材料候选物的基本条件,是可能的含能材料候选物.通过比较各个分子的△E能量值,发现取代后的分子能量差值均高于N2H4(A),由分子前线轨道理论[19-20]可知取代后的分子的稳定性增强.计算所得异构体的总能量与生成热的变化趋势是一致的.当分子总能量越高,其生成热就越大.为进一步研究影响异构体稳定性的因素,还应用NBO方法对它们的超共轭作用进行了计算.通过对比发现,分子的超共轭作用会使体系能量降低,当扣除分子超共轭作用后,体系能量会升高.还发现分子的超共轭作用与甲基的个数成正比,随着甲基数目的增加,分子的超共轭作用能逐渐增大.

表 2 化合物异构体的能量参数

3 羟基(—OH)的取代
3.1 几何性质 图2列出了N2H4的羟基(—OH)异构体,其中(a)为键长(nm),(b)为键临界点处的电荷密度ρ(a.u.),(c)为2ρ(a.u.),(d)为键级.选择以下构型:1-羟基N2H4(G),1,1-二羟基N2H4(H),1,1,2-三羟基N2H4(J)和1,1,2,2-四羟基N2H4(K),来分别讨论取代基的加入对N2H4分子的影响.在这4个构型中H原子逐步被羟基(—OH)所取代.
图2中分别列出了4种羟基(—OH)取代物的键长、AIM和键级分析.从键长数据可以看出,随着羟基(—OH)数目的增多,N—N的键长呈逐渐减小的趋势,电荷密度逐渐增大.其中,1,1,2,2-四羟基N2H4(K)的N—N键长为0.144 9 nm,大于乙氮烷的N—N键长,但经AIM分析发现其2ρ<0,表明所有的化学键仍为共价键.仍然通过NBO方法计算了其键级,发现键级也同样发生了改变.
通过对分子NBO的计算,发现分子中的超共轭作用也与羟基的个数成正比,随着羟基个数增加,分子的离域化能逐渐增大.由表3可知:当分子中的H原子逐渐被取代后,分子中就产生了N原子孤对电子与相邻的N—N键、N—O键、O—H键之间的相互作用.随着取代基数目的增多,二阶稳定化能呈逐渐增大的趋势,且能量普遍高于甲基异构体.—OH取代所产生的超共轭作用能也高于甲基—CH3取代的超共轭作用能.

表 3 化合物异构体的主要二阶稳定化能分析参数
3.2 能量性质 表4列出了计算所得的各异构体的能量参数.可以看出:同样随着取代基数目的增加,总能量逐渐降低.其中二取代物仍有2种:1,1-二羟基N2H4(H)和1,2-二羟基N2H4(I).通过分析能量所得1,1-二羟基N2H4(H)为更稳定的构型.由表2中数据可得当H原子逐步被—OH取代后,分子的总能量与羟基个数呈现很好的线性关系,随着取代羟基数目的增加分子总能量会逐渐降低,每增加一个—OH,分子总能量相应降低75.2 a.u.,比—CH3取代多降低35.9 a.u..比较各个分子的△E能量值同样发现取代后的分子能量差值均高于N2H4(A),且羟基取代的分子能量差△E高于甲基取代的分子,进一步表明羟基取代的产物相比于甲基取代物分子要更稳定一些.生成热作为考察含能物质的一个重要数据,也对它进行了计算,结果表明分子的生成热随着羟基数目的增多呈降低的趋势.—OH取代异构体的生成热低于—CH3取代异构体.通过比较各个分子的△E能量值,发现取代后的分子能量差值均高于N2H4(A),由分子前线轨道理论[17-18]可知取代后的分子的稳定性增强.

表 4 化合物异构体的能量参数
4 结束语
采用密度泛函理论研究了N2H4可能存在的异构体及异构体中的氢原子被甲基和羟基逐步取代后,对原有分子的几何构型、电荷密度、异构体的相对稳定性和生成热造成的影响.通过几何构型优化和振动分析表明,所有异构体均为势能面上的稳定点.通过对AIM及键级分析,发现N—N键长与键临界点电荷密度及键级存在线性关系,研究了其化学键的本质.同时,经过NBO的超共轭作用计算及分子前线轨道理论发现,它们在决定构型稳定性方面均起了重要作用.当引入甲基或羟基后,N原子的孤对电子会与相应的N—C(N—O)键之间发生相互作用,使整个分子的超共轭作用增强.随着取代基数目的增多,总能量和生成热均降低,取代基数目与分子能量的降低值具有很好的相关性.
[1] FOLTZ M F, HOLTZ E V, ORNELLAS D O, et al. The solubilityo-fCL-20 in selected materials[J]. Prop Expl Pyro,1994,19(4):206-295.
[2] FRIED L E, MANAA M R, PAGORIA P F, et al. Design and synthhes is ofenergeticm at erials[J]. Mater Res,2001,31:291-295.
[3] 周世光,吴文健. 高能量密度材料[J]. 化工时刊,1997,11(12):1-6.
[4] 毛双,蒲雪梅,李来才,等. N6H6结构和性质的理论研究[J]. 化学学报,2006,64(14):1429-1436.
[5] 谭英雄. 含能材料分子N6H6及—CH—等电子取代的理论研究[J]. 四川师范大学学报(自然科学版),2009,32(7):998.
[6] GIGUERE P A, LIU I D. On the infraredspectrum of hydrazine[J]. Chem Phys,1952,20(1):136-140.
[7] SCHURATH U, SCHINDLER R N. The photolysis of hydrazine at 206.2 nm in the presence of ethylene[J]. Phys Chem,1970,74(17):188-194.
[8] DAVID W B. High-level ab initio calculations on hydrogen compounds:thermochemistry of tetrazetidine N4H4[J]. Mol Struct:Theo Chem,2002,619(1/2/3):37-43.
[9] DAVID W B. Hartree fock Gaussian -2 and -3 and complete basis set predictions of some thermochemical properties of N4H6[J]. Phys Chem,2001,105(2):465-470.
[10] MY H V, HUYNH D, MICHAEL A, et al. Polyazido high-nitrogen compounds:hydrazo-and-azo-1,3,5-triazine[J]. Angewandte Chemie,2004,43(7):4924-4928.
[11] LI L C, SHANG J. A G3B3 study of N4H4isomers[J]. Mol Struct:Theo Chem,2007,807:207-211.
[12] 郭雅琼,毛双,李强根. N3H3分子取代基效应的量子化学研究[J]. 四川师范大学学报(自然科学版),2014,37(5):703-708.
[13] SYAGE J A, COHEN R B, STEADMAN J. Spectroscopy and dynamics of jet-cooled hydrazines and ammonia. I. Single-photon absorption and ionization spectra[J]. Chem Phys,1992,97(9):6072.
[14] ZEMAN S. Sensitivities of high energy compounds[J]. Struct Bond,2007,125:195-271.
[15] 尚静. 含能材料分子N<,n>H<,n>(n=4~6)系列稳定性分析及其互变异构的理论研究[D]. 成都:四川师范大学,2007.
[16] BIEGLER K F, SCHONBOHM J, DERDAN R, et al. AIM 2000[S]. 2nd ed. Hamilton:McMaster University,2000.
[17] REED A E, WEINHOLD F, CURTISS L A, et al. Natural bond orbital a nalysis of molecular interactions:theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO, and CO2with HF, H2O and NH3[J]. Chem Phys,1986,84:5687-5706.
[18] FRISCH M J, TRUCK G W, SCHLEGEL H B, et al. Gaussian 09[S]. 2nd ed. Wallingford:Gaussian Inc,2009.
[19] CREED D, CALDWELL R A, HIROYUKI O, et al. A frontier molecular orbital rationale[J]. J Am Chem Soc,1977,99:277-278.
[20] HOUK K N. Frontier molecular orbital theory of cycloaddition reactions[J]. Acc Chem Res,1975,8:361-369.
(编辑 周 俊)
Quantum Chemistry Study on the Substituent Effect of N2H4
GUO Yaqiong, GAO Xiaotong, JIA Yalan, ZHANG Yi, MAO Shuang
( 1.CollegeofChemistryandMaterialsScience,SichuanNormalUniversity,Chengdu610066,Sichuan;2.CollegeofArmedPoliceOfficer,Chengdu610213,Sichuan)
Studies on substituent effects of the methyl (—CH3) and hydroxyl (—OH) group on hydrazine were performed. For derivatives substituted methyl, the N—N bond length of hydrazine was increased, and charge dengsity was lessened. For derivatives substituted hydroxyl, the N—N bond length of hydrazine was shortened. By NBO (nature bond orbit), the bond order of N—N bond was decreased with the increase of the number of the substituent. The hyperconjugation played a important role in the relative stability of isomers. The hyperconjugation from the lone pair electrons of atom N to the N—C (N—O) bond was increased by the introduction of substituent. The total energy and formation heat were decreased with the addition of substituents. A good correlation was found between the numher of substituent and molecular relative stability.
quantum chemsity; substituent effect; hydronitrogen; isomer; relative stability
2015-06-01
四川省教育厅自然科学重点基金(10ZA011)
O641
A
1001-8395(2017)01-0101-05
10.3969/j.issn.1001-8395.2017.01.017
*通信作者简介:毛 双(1972—),男,副教授,主要从事物理化学的研究,E-mail:wlhxms@sohu.com
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
数学物理学报(2022年1期)2022-03-16
云南化工(2021年8期)2021-12-21
数学物理学报(2021年6期)2021-12-21
河北理科教学研究(2020年1期)2020-07-24
应用数学(2020年2期)2020-06-24
青岛大学学报(工程技术版)(2019年2期)2019-09-10
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
