
UGT1A1*6纯合突变型Gilbert综合征伴重度高胆红素血症1例报道并文献复习
2020-02-18 03:25刘瑶陈娅陈鸿邱隆敏
肝脏 2020年1期
刘瑶 陈娅 陈鸿 邱隆敏
患者,女,19岁,因“全身皮肤黄染15年,加重伴上腹不适1 d”入院。既往全身皮肤、巩膜黄染15年,多次因皮肤巩膜黄染加深入住当地县医院,病因未明。入院查体:皮肤、巩膜重度黄染,上腹部压痛,肝脾未及,余无特殊。上腹部CT平扫+增强提示:急性胰腺炎。血清淀粉酶:1 267 U/L;血常规:白细胞总数 11.42×109/L,中性粒细胞绝对值 10.85×109/L,血红蛋白102.0 g/L,红细胞总数 3.12×1012/L,血小板总数 199×109/L;肝功能:ALT 231 U/L,AST 246 U/L,GGT 548 U/L,TBil 655.6 μmol/L,DBil 240.3 μmol/L,IBil 415.3 μmol/L。经积极治疗后腹痛症状好转,但黄疸未消退,复查肝功能示:ALT 81 U/L,AST 108 U/L,TBil 306.9 μmol/L,DBil 95.1 μmol/L,IBil 211.8 μmol/L。完善相关辅助检查:铜蓝蛋白:29.3 mg/dL,微量元素:铜11.2 μmol/L,甲状腺功能、网织红细胞计数、自身免疫性抗体、病毒四项、HBV血清标志物、丙肝抗体、甲戊肝抗体均未见异常,故除外病毒性肝炎、溶血性黄疸、梗阻性黄疸、铜蓝蛋白血症、肝豆状核变性、自身免疫性肝病。
结合患者黄疸病史长达15年,一般情况好,病情未进行性加重,血清胆红素以非结合胆红素升高为主等特点,考虑先天性非溶血性黄疸可能性大,为进一步确诊,行UGT1A1基因测序检查,结果提示位于Exon1的错义突变:UGT1A1*6(纯合),该突变与葡萄糖醛酸转移酶活性降低有关,见图1。

图1 患者UGT1A1的检测结果
为进一步明确该基因的遗传特点,在获得患者及家属同意后,抽取其父母外周血进行UGT1A1基因测序检查。结果提示父亲UGT1A1基因存在变异,位于Exon1的错义突变,c.211G>A,即UGT1A1*6(杂合),见图2;母亲UGT1A1基因存在变异,位于Exon1的插入突变UGT1A1*28(杂合)和错义突变UGT1A1*6(杂合),见图3。

图2 患者父亲UGT1A1的检测结果
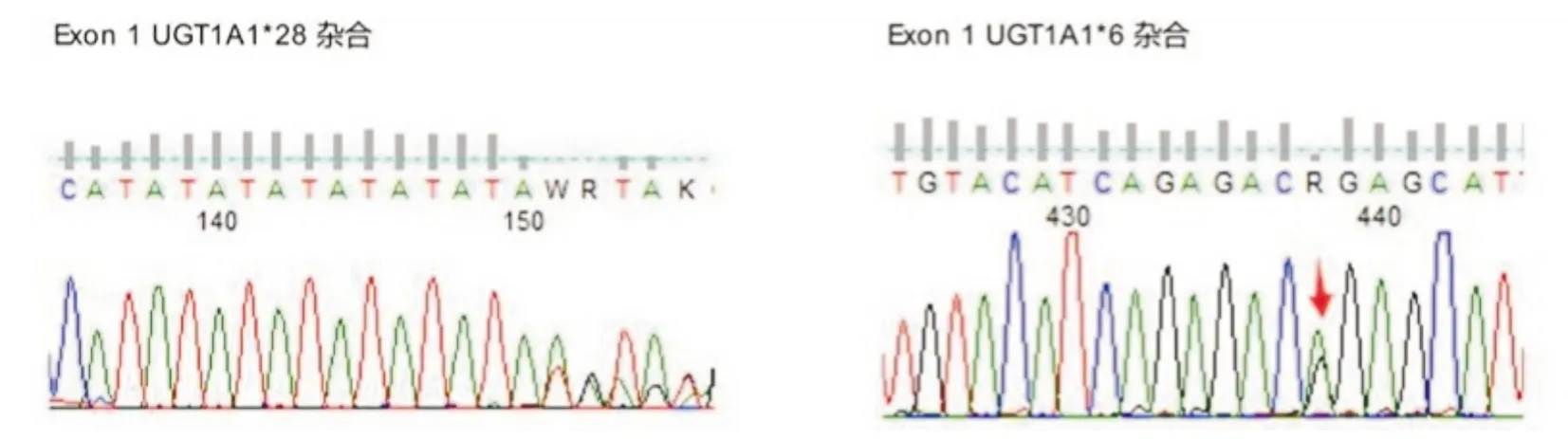
图3 患者母亲的UGT1A1的检测结果
根据基因检测的结果,该患者确诊为Gilbert综合征(Gilbert Syndrome,GS)。本例Gilbert综合征存在UGT1A1基因Exon1的错义突变,该突变为UGT1A1基因第1外显子第211位的鸟嘌呤(G)突变为腺嘌呤(A),为Gly71Arg纯合突变,符合常染色体隐性遗传方式。
为进一步明确该病的特点,我们对CNKI、Wanfang、Pubmed、Embase数据库近10年相关文献进行检索,语言为中英文,中文检索词为“Gilbert综合征”、“吉尔伯特综合征”、“日尔贝综合征”、“Gilbert病”,英文检索词为“gilbert syndrome”、“gilbert disease”、“GS”。采用纳入与排除标准:①患者经基因检测确诊为GS,且相关基因突变类型为UGT1A1*6或UGT1A1*28;②有完整的临床资料,包括年龄、性别、实验室检查(血清胆红素)等资料;③排除其他基因突变类型的GS、重复病例报道、综述以及资料不完整的文献。最终纳入12篇文献,共收集12例GS患者。回顾性分析患者的病例资料,结果见表1。

表1 12例GS患者基本信息与突变基因类型
12例GS患者中,男7例,年龄19~67岁,平均年龄37.3岁;女5例,年龄22~64岁,平均年龄41.8岁。12例GS患者平均总胆红素140.3 μmol/L,平均非结合胆红素115.9 μmol/L。其中4例GS患者为UGT1A1*6突变型,4例GS患者为UGT1A1*28突变型,4例GS患者合并有UGT1A1*6及UGT1A1*28的共同突变。2例患者合并遗传性球细胞增多症(HS),2例患者合并Crigler-Najjar综合征(CNS),1例合并原发性胆汁性胆管炎(PBC),1例合并Dubin-Johnson 综合征(DJS),1例合并HBV感染,1例合并骨髓增殖性肿瘤(MPN)。12例患者的主要临床表现均以黄疸为主,当GS合并DJS或CNS为双重遗传性黄疸,除1例[11]因胆囊结石行胆囊切除术,1例[3]行苯巴比妥诊断性治疗,其余患者针对GS疾病本身无特殊治疗。
讨论Gilbert综合征是一种遗传性非溶血性黄疸,其发病机制主要与UGT1A1的基因突变有关,当UGT1A1的基因发生突变导致尿苷二磷酸葡萄糖醛酸转移酶的表达和功能下降甚至缺失[13],使胆红素结合缺陷致非结合胆红素水平升高,从而导致以非结合胆红素升高为主的高胆红素血症[14]。根据既往的报道[15],UGT1A1基因突变的频率在不同种族之间存在差异,其中较为常见的为UGT1A1第1外显子TATA-box的插入突变和错义突变,前者表现为A(TA)7TAA(又称UGT1A1*28)序列取代正常的A(TA)6TAA,而后者表现为第1外显子中G71Arg突变(G71R;UGT1A1*6),即UGT1A1基因第1外显子第211位的鸟嘌呤(G)突变为腺嘌呤(A)[13]。通过检索近10年的相关文献及结合本次病例报道发现GS临床症状多以单纯黄疸为主要表现,为轻度的血清胆红素升高,同时Saki等[16]的研究也表明,GS疾病本身很少引起严重的高胆红素血症,但当其合并其他病症可加重高胆红素血症。本文献复习中报道的中重度血清胆红素升高患者均有其他合并症的存在,比如CNS、HS、PBC、HBV等。本例患者血清总胆红素高达655.6μmol/L,在除外其他合并症可能的前提下,考虑为胰腺炎症因素加重其高胆红素血症。
综上所述,Gilbert综合征是一类与遗传相关的以非结合胆红素升高为主的高胆红素血症,临床主要散发性无症状的黄疸为特征[17],其诊断的金标准为对UGT1A1突变基因进行检测,而通常针对原发疾病本身无特殊治疗。由于该病起病隐匿,症状多轻微,临床易漏诊、误诊,因此对于该病的诊断及基因的确定可以避免不必要的检查以加重患者的心理负担及经济负担。目前随着基因检测技术的发展,对于该病的诊断更为明确,但是由于我国目前尚缺乏对基因样本的大数据研究,本次报道的1例UGT1A1*6(纯合)突变Gilbert综合征,可为该病的诊断及基因突变分型提供相关病例资料。
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
中国生殖健康(2020年2期)2021-01-18
中南医学科学杂志(2019年6期)2019-12-05
浙江中医杂志(2019年3期)2019-01-05
祝您健康·文摘版(2018年6期)2018-10-21
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
护士进修杂志(2017年2期)2017-02-16
蚌埠医学院学报(2016年7期)2016-09-01
