
A Chemical Kinetics Perspective on the High-Temperature Oxidation of Methane and Propane through Experiments and Kinetic Analysis
2022-02-01 04:02MaShoutaoYangZheZhuYunfengSunBingJiangJieXuWeiMengRuiji
中国炼油与石油化工 2022年4期
Ma Shoutao; Yang Zhe; Zhu Yunfeng; Sun Bing; Jiang Jie; Xu Wei; Meng Ruiji
(State Key Laboratory of Safety and Control for Chemicals, SINOPEC Research Institute of Safety Engineering, Qingdao Shandong 266101, China)
Abstract: In the conversion of methane and propane under high temperature and pressure, the ignition delay time (IDT) is a key parameter to consider for designing an inherently safe process. In this study, the IDT characteristics of methane and propane (700–1000 K, 10–20 bar) were studied experimentally and using kinetic modeling tools at stoichiometric fuel-tooxygen ratios. All the experiments were conducted through insentropic compression. The reliable experimental data were obtained by using the adiabatic core hypothesis, which can be used to generate and validate the detailed chemical kinetics model. The IDTs of methane and propane were recorded by a rapid compression machine (RCM) and compared to the predicted values obtained by the NUIGMech 3.0 mechanism. To test the applicability of NUIGMech 3.0 under different reaction conditions, the influence of temperature in the range of 700–1000 K (and the influence of pressure in the range of 10–20 bar) on the IDT was studied. The results showed that NUIGMech 3.0 could reasonably reproduce the experimentally determined IDT under the wide range of conditions studied. The constant volume chemical kinetics model was used to reveal the effect of temperature on the elementary reaction, and the negative temperature coefficient (NTC) behavior of propane was also observed at 20 bar. The experimental data can serve as a reference for the correction and application of kinetic data, as well as provide a theoretical basis for the safe conversion of low-carbon hydrocarbon chemicals.
Key words: ignition delay time, NUIGMech 3.0, negative temperature coefficient, kinetic analysis
1 Introduction
The preparation and conversion of low-carbon hydrocarbons, such as methane and propane, are important processes in the chemical industry. In recent years, many processes involving the mixing and reaction of oxygen and combustible gas are performed under high temperatures and pressures with the development of technology. The output of natural gas increases every year owing to the massive exploitation and application of shale gas, and the cost of low-carbon hydrocarbon gases such as methane and propane significantly reduced. Therefore, the economic benefits of some new technologies, such as the natural gas to olefin process, are guaranteed[1-2].
Methane and propane are important components of natural gas and liquefied petroleum gas, respectively, and they are also typical representatives of low-carbon hydrocarbons. Studying the ignition delay behavior of methane and propane at stoichiometric ratios can provide new insights into their oxidation kinetics and offer an experimental basis for the establishment of kinetic models. Ajoy et al.[3]studied the ignition delay time (IDT) of propane at different temperatures. In addition, Goldsborough et al.[4]and Sabia et al.[5]reviewed the ignition behavior of propane. Baigmohammadi et al.[6-7]have studied the IDT characteristics of pure hydrocarbons (C1–C2), including methane (CH4), ethane (C2H6), and ethylene (C2H4), and their binary mixtures. Subsequently, Martinez et al.[8]extended this work to higher hydrocarbons by studying the IDT of binary mixtures of ethylene (C2H4)/propane (C3H8) and C2H6/C3H8in a shock tube and rapid compression machine (RCM) under a wide range of temperatures, pressures, equivalence ratios, and dilutions. Although many researchers have measured the IDTs of CH4and C3H8, there is still a lack of mechanistic data for CH4and C3H8at stoichiometric ratios, except for the recent research on different liquified petroleum gas (LPG) fuels for vehicles by Ramalingam[9].
On the other hand, although the energy spent on prototype design and testing may be reduced with the development of computing power and simulation technology, differences exist between the simulation and experimental results. Therefore, developing an accurate chemical kinetics mechanism to predict the conditions related to different applications is extremely important. In addition, the emission and ignition behavior of different fuels can be understood through chemical kinetics mechanisms. Understanding the oxidation process of these fuels at the basic level is helpful to further understand the accurate prediction of their reactivity. Although low-carbon hydrocarbons have been studied[6-8], kinetic data on fuel mixtures at application-related pressures are lacking.
Nevertheless, IDTs measured experimentally at different pressures and temperatures can help researchers develop more complex and high-fidelity chemical kinetics models. This can also result in a valuable database, which can be used to modify the IDT characteristics of liquified natural gas mixtures. This database can also be widely used in industrial furnaces and internal/external combustion engines operating under the homogeneous charge compression ignition, exhaust gas recirculation, and moderate or severe low oxygen dilution combustion modes[8-9].
The main objective of this study is to experimentally determine the IDTs of CH4and C3H8under different pressures and temperatures to propose a new improved method for analyzing the combustion and explosion mechanisms. The results can represent the spontaneous combustion behaviors of CH4and C3H8under a wide range of temperatures and pressures. The development of the mechanism is an evolving process, and the experimental work conducted in this study and in previous research contribute to the development of NUIGMech3.0[9-10]. Existing studies on LPG fuels for vehicles[9]and C3H8[10]show that the mechanisms available in the literature cannot capture the reactivity of pure components and their mixtures in a consistent manner.
In summary, this study is divided into three parts: (I) designing new experiments under a wide range of operating conditions based on the principle and method of isentropic compression; (II) experimental measurements; (III) simulation and chemical kinetics analysis using the NUIGMech 3.0 mechanism through the CHEMKINPRO simulation software. The supporting information document contains the RCM-simulated nonreactive traces, original spreadsheet of the experimental tests, and low-/high-pressure shock tube oscilloscope traces. In addition, general information about the gases (fuel/oxygen/argon/nitrogen), experimental facilities, and data acquisition system can also be obtained from this document.
2 Experimental
2.1 Experimental device
The IDT measurements at low and medium temperatures were obtained in a heated RCM, and the RCM device is shown in Figure 1. It is mainly composed of three parts: pneumatic drive part, moving piston brake section, and combustion reaction chamber. A typical creviced piston is used to suppress the eddy current caused by the boundary layer in the rapid compression process to ensure a uniform thermodynamic state after compression. In this study, the compression temperature (TC) and compression pressure (PC) were obtained at the end of compression (EOC), which was determined by adjusting the compression ratio (CR), initial temperature (T0), and initial pressure (P0). The dynamic pressure was obtained by the piezoelectric pressure sensor (Kistler 6125b) and charge amplifier (Kistler 5015). TheTCcannot be measured directly; therefore, it is calculated according to the adiabatic core hypothesis.[11]

whereP0andT0are the initial pressure and initial temperature, respectively. γ refers to the temperaturedependent specific heat ratio of the reactive mixtures.The reliability of the independently designed RCM was verified through repetitive experiments, and the results are shown in Figure 2. The IDTs of 16.7%CH4/33.3%O2/50%Ar (mole fraction) are consistent at 900 K and 20 bar, indicating that the device has some stability. As shown in Figure 3, the total IDT in this study is defined as the time span from EOC to the maximum of the pressure derivative. Here, the reported representative value is close to the average value. The uncertainty of the IDT in the current work mainly comes from the initial state and nonideal compression process, which is estimated to be less than ±15%.
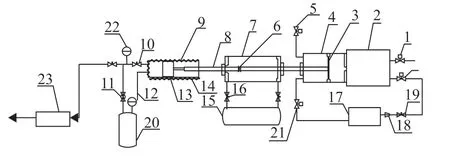
Figure 1 Rapid compression machine
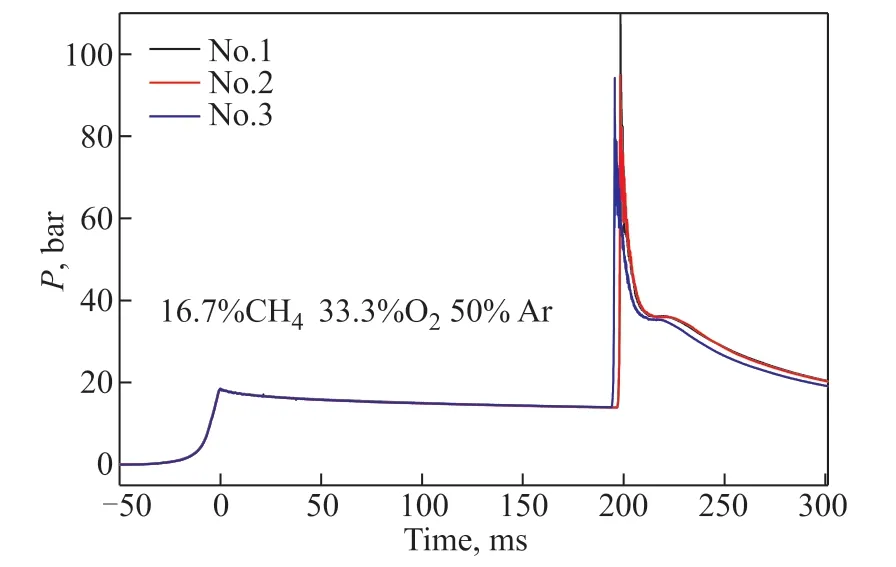
Figure 2 Reliability of RCM
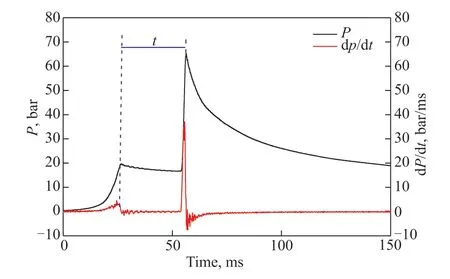
Figure 3 Definition of ignition delay time using RCM
2.2 Test fuel and conditions
High-purity CH4(99.99%) and C3H8(99.999%) were provided by Dalian Date Co., Ltd. To ensure the accuracy of IDTs, the purity of nitrogen and oxygen was above 99.99%. Before the experiment, the pressure of the premix tank was adjusted to 3 Pa using a vacuum pump, and high-purity nitrogen was used for repeated replacement. Next, the mixture under the test scheme was configured according to the volume ratio. To ensure complete mixing of the mixture, the premix tank was kept stationary for more than 4 h after feeding. Table 1 presents the test conditions and the compositions of the low-carbon hydrocarbons, oxygen, and nitrogen. As the mixing proportion is according to the volume ratio, the mole fraction of the alkane corresponds to the volume fraction of the fuel in the mixture.
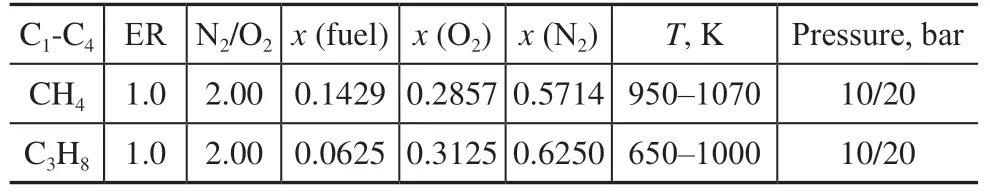
Table 1 Test conditions and mixture compositions.
3 Simulation Specifications
3.1 Simulation methods
CHEMKIN-PRO was used for calculating the IDTs because it requires less computational resources. The input files of CHEMKIN-PRO include the gas phase dynamics, surface dynamics, thermodynamic, and component transport files. The gas phase dynamics and thermodynamics files are used in the calculation process. The gas phase dynamics document contains the elementary reaction parameter and the corresponding reaction rate constant (k(T)). In general, the reaction rate constantK(T) is expressed as follows:

whereAis the pre-exponential factor,bis the temperature index, andEais the activation energy. For pressuredependent reactions, (+M) generally exists in the elementary reaction formula. The chemical reaction rate constant is a function of temperature and pressure. The mechanism file used in this study was that of the NUIGMech 3.0 mechanism, which could be directly imported into CHEMKIN-PRO. In addition, the thermodynamic document included thermodynamic data such as the specific heat capacity, entropy, and enthalpy of components. These were obtained by polynomial calculations, and the details are available in the CHEMKIN-PRO tutorial manual.
In this study, the NUIGMech3.0 mechanismand nonreactive input files were used for the IDT simulation, and the real reaction temperature could be determined in the nonreactive experiment, which was carried out by replacing oxygen with nitrogen. The obtained nonreactive pressure trace could be transformed into the effective volume trace using the isentropic relationship, and the effective volume trajectory and initial experimental conditions were used to perform zero-dimensional (0-D) simulations[12]. The IDT simulation, calculation of the species mole fraction distribution, as well as sensitivity and yield analyses are carried out using an internal script developed by Can-tera[13]. The IDT simulation also provided the change trace of the species during IDT; the species formation profile was used for comparison with the experimental data and the normalization process was also realized. Existing studies[10]discuss the calculation methods for species formation profiles, with no further details. Constant volume reactors can be used for sensitivity and production rate analyses to determine the most sensitive reactions and pathways leading to key intermediates controlling fuel reactivity.[14]There are two models for the IDT simulation. The first is the constant volume model without considering nonideal facility effects, such as the heat loss during compression in the RCM. The CONV model is also used for dynamic analysis of engines to save calculation time. Studies[15]report that the predicted IDT is the time required for increasing the initial temperature by 400 K, and the IDT based on the temperature standard is close to the time derivative of the pressure or OH radical concentration. The second is called the variable volume model (i.e., volume as a function of time)[16], and the heat loss from the RCM in the model can be considered for the IDT simulation. Nativel et al.[17]have used the pressure increase (dp/dt) for the IDT simulation, and it reveals the dependence of dp/dton the initial pressure, inner diameter, Mach number, and specific heat ratio over a wide range of conditions[18]. Therefore, the adiabatic equation and ideal gas law obtained through the variable volume–time profiles are used for processing the RCM experimental data. The variable volume–time profiles are calculated from the nonreactive pressure trace. Figure 4 shows a typical volume trajectory and it represents a rapid compression process, followed by a slow isentropic expansion process. The definition of the predicted IDTs is analogous to that determined experimentally. All volume traces are provided in the Supplementary Material.

Figure 4 Conversion of nonreactive pressure traces to volume traces. solid lines: volume profiles and dashed lines: pressure profiles
3.2 Brute-force sensitivity analysis
The 0-D homogeneous constant volume reactor model in CHEMKIN-PRO was used to simulate the premixed combustion process of CH4/C3H8and oxygen/nitrogen, and the simulated reaction path results helped determine the IDT of CH4or C3H8and reveal the negative temperature coefficient (NTC) behavior of C3H8. Although sensitivity analysis determines the influence of small disturbances on the system characteristics by analyzing the relationship between a single reaction and the IDT in the chemical kinetics model, the IDT (τ) is perturbed by direct changes in the pre-exponential factor in the Arrhenius equation. Sensitivity analysis coefficients can be either positive or negative, with a positive value corresponding to a reaction that inhibits reactivity, giving a longer IDT, and vice versa. It is also possible to evaluate which reactions in the mechanism have the greatest impact on the target parameters and variables. The formula for calculating the sensitivity coefficient of the IDTs to the elementary reaction rate is as follows[19]:

whereSiis the sensitivity coefficient andτis the IDT.kirepresents the rate constant of theithelementary reaction, whereas 2kirepresents the rate constant of the elementary reaction multiplied by 2.Sicharacterizes the influence of the pre-exponential factor of the elementary reaction on the IDTs, and OH, which is the characteristic functional group in ignition, can be selected as the indicator to measure the effect of the elementary reaction rate on the overall reaction rate.
4 Result and Discussion
4.1 Performance of NUIGMech3.0 and the corresponding correlations versus experimental data
Figure 5 compares the performance of the NUIGMech3.0 mechanism with the new experimental RCM data of the CH4/C3H8fuel mixture at different temperatures. Figure 5 shows that the NUIGMech3.0 mechanism can predict all the measured IDTs under random distribution conditions. The results also show the effect of temperature on the IDTs. For the CH4system, the NUIGMech3.0 mechanism underestimates the IDTs when the temperature exceeds 1000 K. However, for the C3H8system, the NUIGMech 3.0 mechanism can reliably predict the IDT within the measured temperature range, but the predicted value is smaller than the experimental value. In addition, the NTC behavior of experimental data are more obviouse than that of the predicted data. The difference between the simulation and experimental results is mainly caused by the deviation in the model structure and uncertainty of the experimental data.The oxidation reaction mechanism considered in this study contains the energy barriers and rate constants of all elementary reactions, which can describe the oxidation process of fuel to final products. The method of optimizing the rate constants based on experimental data has been applied to the small molecule mechanisms owing to the lack of reliable methods for determining the rate constants of most elementary reactions.This is because all the chemical reaction kinetics mechanisms are constructed based on the Arrhenius equation, and the reaction rate of each elementary reaction is also determined by the Arrhenius equation.Thus, the preliminary construction of the chemical reaction kinetics mechanism can be completed by combining the thermodynamic and transport parameters of each component. CHEMKIN-PRO is used to simulate the IDTs under different working conditions, and the kinetic mechanism of the chemical reaction is also corrected based on the difference between the experimental and simulation data.Consequently, high-precision quantum chemical calculationswere performed to modify the preexponential factor and activation energy of the elementary reaction, which could enhance the mechanism document and improve the accuracy of the simulation results.
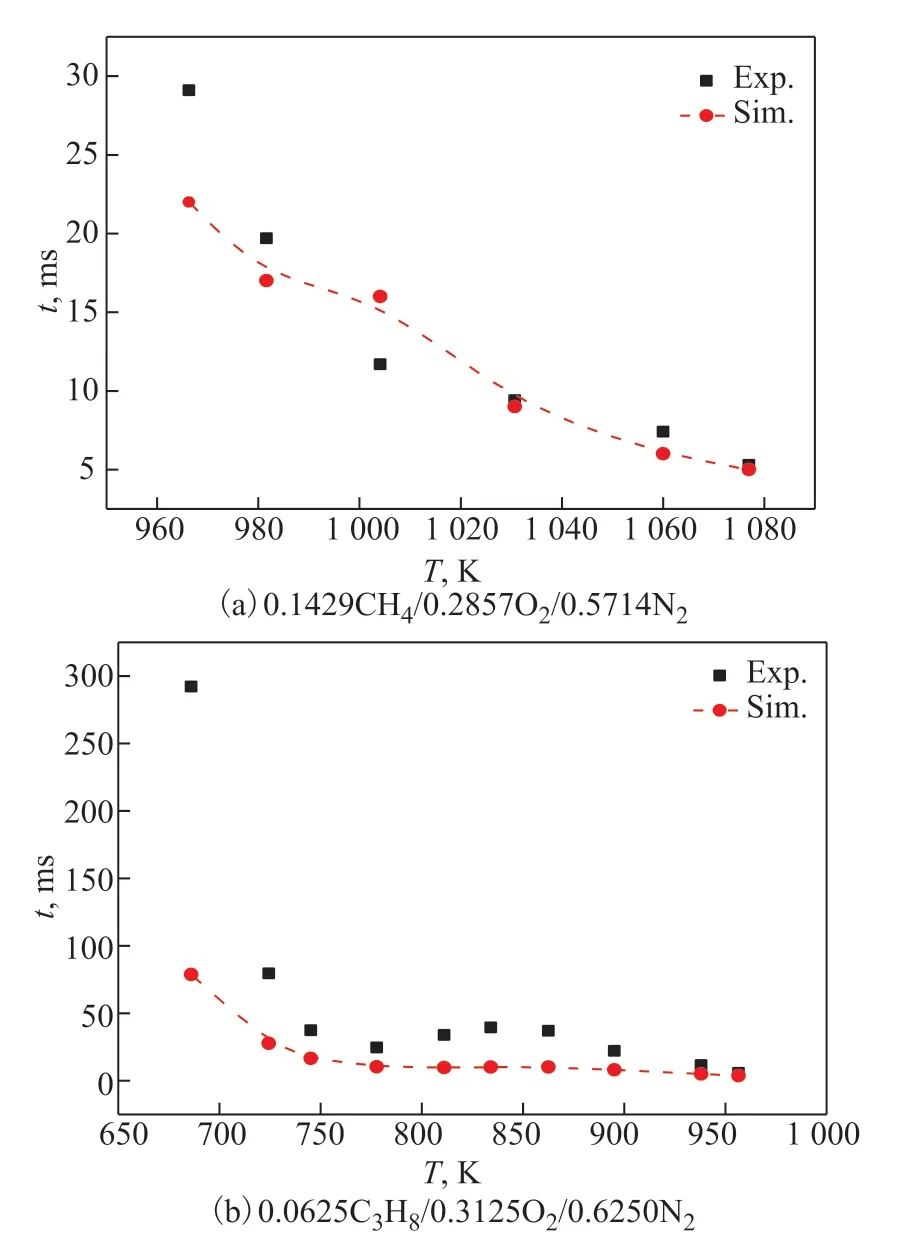
Figure 5 Experimental and simulated IDT data of CH4/C3H8 mixtures
4.1.1 Effect of pressure
As observed in Figure 6(a) and (b), CH4and C3H8do not ignite automatically under 10 bar in the entire temperature range. However, under 20 bar, the starting ignition temperature and IDT of CH4are approximately 950 K and 62 ms, respectively, and those of C3H8are 694 K and 292.2 ms, respectively. The IDTs increase as the temperature increases for CH4or C3H8. When the temperature rises to 794 K, the IDT of C3H8is 33.8 ms, and it is 39.5 ms when the temperature rises to 825 K. This ignition behavior of C3H8is called NTC behavior. The ignition delay behavior of CH4and C3H8does not occur autonomously under 10 bar but occurs under 20 bar because increasing the pressure at a given temperature means increasing the local reactant concentration. Increasing the pressure increases the collision frequency among fuel molecules.
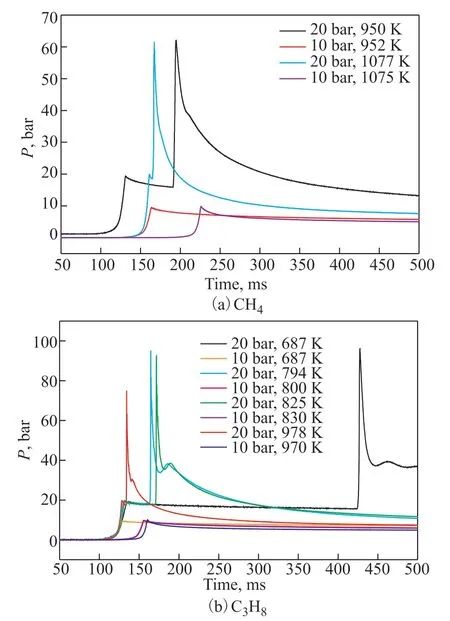
Figure 6 IDTs of CH4 and C3H8 at different temperatures and pressures
In contrast, according to Jost[20], the IDTs are expected to be inversely proportional to the number of carbon atoms in the molecule under the same initial pressure and temperature conditions. The expected behavior of CH4and C3H8shows that the reaction activity of CH4is much lower than that of high-carbon hydrocarbons. The main reason is that CH4does not have a C–C bond, and its spontaneous combustion behavior is a free radical reaction caused by the fracture of the C–H bond. The self-ignition condition of CH4is more difficult compared to other alkanse. The reason is that the energy required to breakage the C–H bond is greater than that required to breakage the C–C bond, and activation energy of elementary reactions in hydrocarbon cracking for CH4is also higher than that of other alkanes.
4.1.2 Sensitivity analysis
To further study the main elementary reactions of ignition delay, CHEMKIN-PRO was used for sensitivity analysis. Figure 7(a) shows the sensitivity analysis results for the OH group in 0.1429CH4/0.2857O2/0.5714N2at 20 bar and 966 K, in which the negative value indicates that the increased elementary reaction rate constant promotes the formation of the OH group and reduces the IDTs. Figure 7(a) shows that the elementary reactions that promote the IDTs of CH4are CH4+HO2=CH3+H2O2and CH4+CH3O2=CH3+CH3O2H, among others. The free radicals involved in the elementary reaction are OH and CH3. The elementary reactions that inhibit the IDTs of CH4are 2CH3+(M)=C2H6+(M) and H+O2+(M)=HO2+(M). Although H2O2generates two OH groups, which can increase the OH group exponentially and significantly accelerate the ignition process, CH3, which is a free radical with reduced reaction activity, forms easily in the chain termination reaction CH3+HO2=CH4+O2at low temperatures. In addition, CH3+HO2=CH3O+OH consumes CH3radicals and generates OH radicals, which can significantly promote the ignition process. Figure 7(b) shows the sensitivity analysis results of the OH radical at a high temperature, and the elementary reactions that promote the IDTs of CH4are CH3+HO2=CH3O+OH, CH4+HO2=CH3+H2O2, CH3+O2=CH2O+OH, and CH3O2+CH3=2CH3O. The elementary inhibition reaction of the CH4ignition delay is similar to that at a low temperature, which consumes free radicals, and the final reaction produces CH4and C2H6. The generated CH4and C2H6are stable products, which are not conducive to ignition. The key step of the reaction is mainly CH4cracking to generate CH3at high temperatures; H and O react to generate OH. CH3is more active at high temperatures, and it can react with HO2to generate OH, thus initiating chain transfer.

Figure 7 Sensitivity coefficients for IDTs of CH4 at different temperatures
Figure 8 shows the sensitivity coefficients for the IDTs of C3H8at different temperatures, indicating whether the elementary reaction can inhibit or promote ignition. Figure 8(a) shows the sensitivity coefficient of 0.0625C3H8/0.3125O2/0.6250N2at 20 bar and 687 K. The elementary reactions that promote the IDTs of C3H8are C3H8+OH=NC3H7+H2O, IC3H7O2=C3H6OOH2-1, NC3H7O2=C3H6OOH1-3, and C3KET13=CH2O+CH2CHO+OH. The elementary reactions that inhibit the IDTs of C3H8are C3H8+OH=IC3H7+H2O, IC3H7O2=C3H6+HO2, and NC3H7O2=C3H6+HO2. The ignition of C3H8involves the extraction of the H atom by HO, resulting in the dominance of the isopropyl radical pathway. Figure 8(b) shows the sensitivity coefficient of 0.0625C3H8/0.3125O2/0.6250N2at 20 bar and 824 K. The elementary reactions that promote the IDTs of C3H8are NC3H7O2=C3H6OOH1-3, C3H6OOH2-1O2=C3KET21+OH, and IC3H7O2=C3H6OOH2-1, among others. The elementary reactions that inhibit the IDTs of C3H8are IC3H7O2=C3H6+HO2, C3H6+HO2=C3H6OOH2-1, NC3H7O2=C3H6OOH1-2, and 2HO2=H2O2+O2. In addition, Fig 8(c) shows that the chain branching reaction does not produce the OH radical; however, the formation of the OH radical in the chain branching reaction path will lead to NTC behavior at low temperatures. The main reason is that the reaction rate of H2O2+(M)=2OH+(M) at low temperatures is higher, which leads to accelerated reaction rates of the OH-related elements, resulting in the chain transfer reaction. Therefore, the competition between chain branching and chain transfer is the fundamental reason for the NTC behavior of alkanes. Figure 8(c) shows the sensitivity coefficient of 0.0625C3H8/0.3125O2/0.6250N2at 20 bar and 948.5 K. The elementary reactions that promote the IDTs of C3H8are C3H8+HO2=IC3H7+H2O2, C3H8+HO2=NC3H7+H2O2, CH3O2+C3H8=CH3O2H+IC3H7, and H2O2(+M)=2OH+(M). The elementary reactions that inhibit the IDTs of C3H8are 2HO2=H2O2+O2and C3H6+HO2=C3H6OOH2-1. When the chain branch C3H7is generated at high temperatures, H2O2is also generated, and H2O2further generates 2OH, leading to chain transfer. Therefore, when the temperature exceeds 900 K, H2O2is basically consumed in the production process. This reaction is the most important one to determine the ignition delay in the range of 800–1000 K.
4.2 Reaction path analysis
4.2.1 Methane
As the simplest low-carbon hydrocarbon, the combustion mechanism of CH4is relatively simple. Figure 9 shows the reaction path analysis of CH4at different temperatures. CH4first reacts with OH/H and O radicals to produce simple CH3radicals and then reacts with HCO to produce CH3CHO. HCCO reacts with OH radicals to produce CO, and finally, CO reacts with OH/HO2to produce CO2. The reaction path of CH4at the high temperature is CH4→CH3/CH3O2→CH2(S)→CH/HCO→CO2. In conclusion, the elementary reaction of hydrogen cracking is the main step of chain branching initiation for the CH4 system. The energy required for hydrogen cracking is high, which leads to low activity of CH4 and high energy for self-ignition.
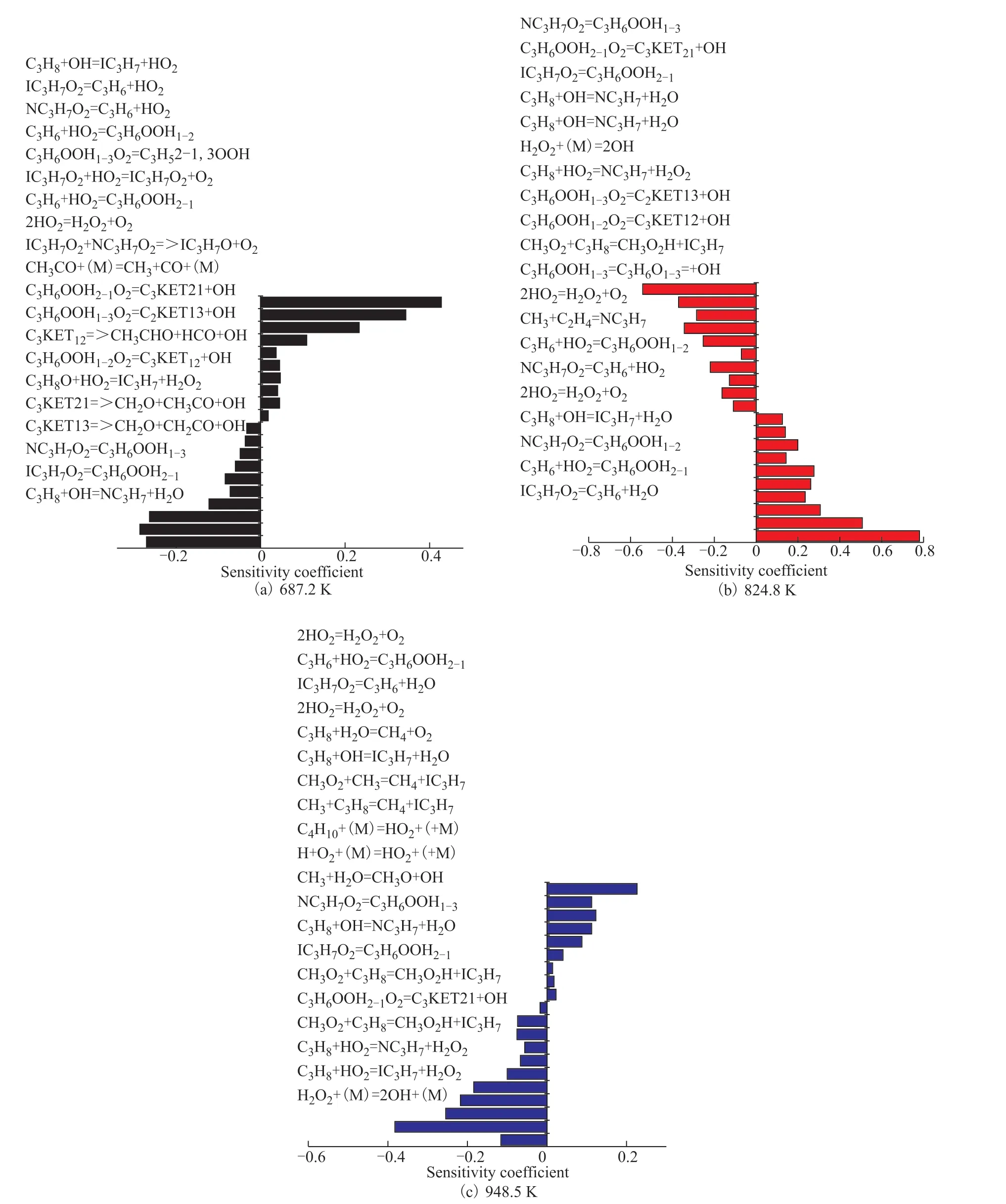
Figure 8 Sensitivity coefficients for IDTs of C3H8 at different temperatures

Figure 9 Rate of production analysis using NUIGMech3.0 mechanism for CH4 under different conditions
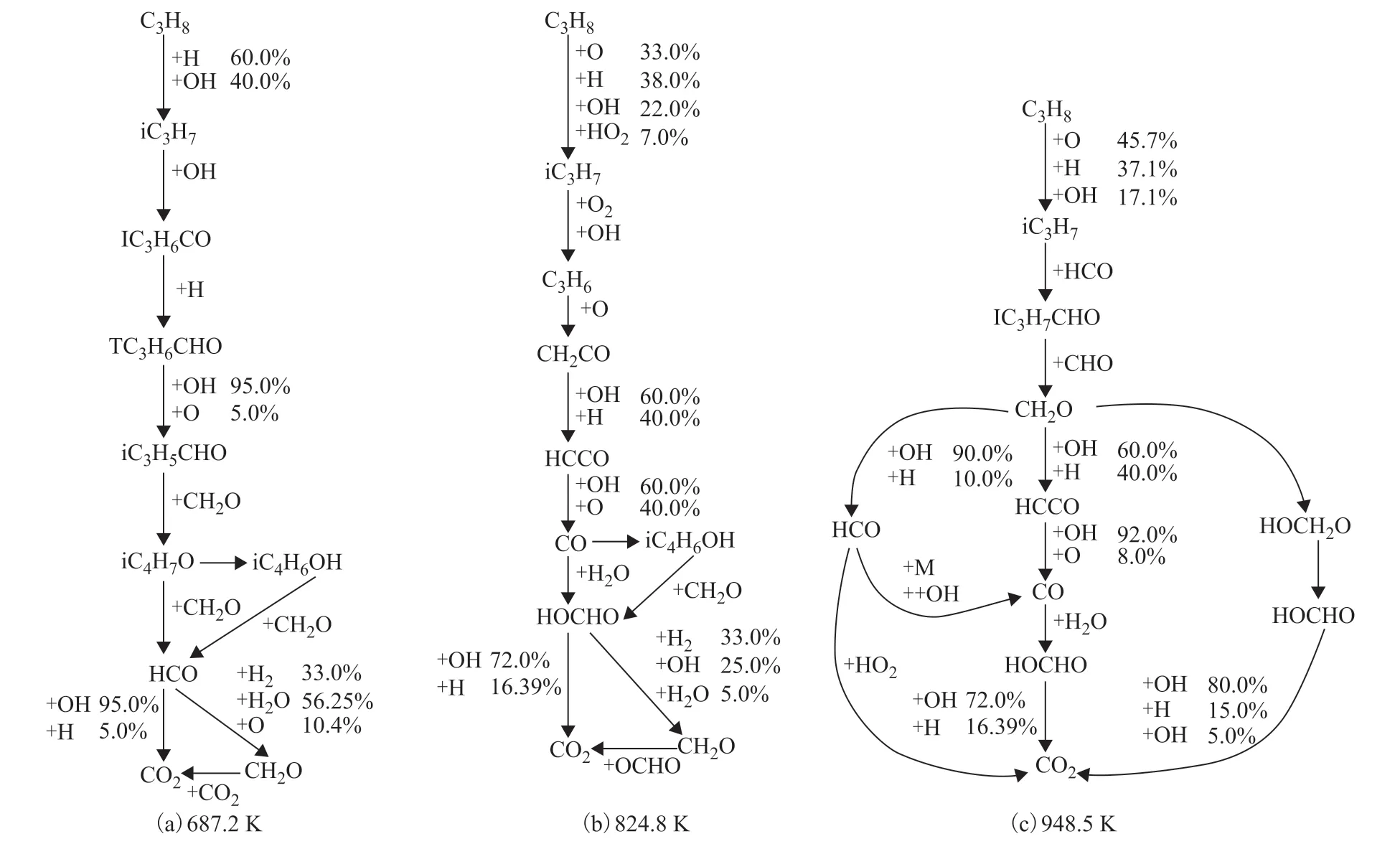
Figure 10 Rate of production analysis using NUIGMech3.0 mechanism for C3H8 at different conditions
4.2.2 Propane
Burke et al.[21-22]underestimated the reactivity of C3H8. The NUIGMech3.0 mechanism can well predict the NTC behavior of C3H8,and the predicted trend is basically consistent with the experimental data. This section mainly discusses the C3H8reaction path at different temperatures, and the results are shown in Figure 10. The consumption of C3H8is for the extraction of H atoms by OH, which leads to the dominance of isopropyl radicals at the studied conditions. The formation of propylene is central, which is mainly produced by the synergistic elimination of the propyl peroxy radical in the n-propyl and isopropyl radical pathways. However, the isopropyl radical pathway is dominant. C3H8branching does not play a major role at low temperatures, and the HOCHO chain branching reaction does not produce OH radicals. Nevertheless, it consumes active OH and HO2radicals at medium temperatures, resulting in NTC behavior. In addition, acetaldehyde is another important intermediate produced in the oxidation process of C3H8, and it is mainly formed through isopropoxy and β fracture. The branching ratio of the propyl radical has been studied earlier[23-24]. Therefore, the pressure-related synergistic elimination reaction may be the reason for the overprediction of C3H8at 20 bar.
5 Conclusion
In this study, an RCM was used to determine the IDTs of CH4and C3H8in the low and medium temperature ranges under pressures of 10 and 20 bar. At 10 bar, there was no spontaneous combustion of CH4and C3H8within the measured temperature range. CH4and C3H8exhibited ignition delay behavior at 20 bar, and C3H8showed obvious NTC behavior. This work is an extension of a previous study on CH4/C3H8oxidation, where the speciation technique was elaborated. The NUIGMech3.0 mechanism could predict the IDTs of CH4and C3H8within the temperature range, but the predicted value was lower than the experimental value, and the NTC behavior of C3H8was not obvious. In addition, NUIGMech3.0 was selected for the sensitivity and reaction path analyses of the OH radical for CH4and C3H8systems at different temperatures, which revealed the reaction mechanisms of the ignition delay process of CH4and C3H8. For CH4, dehydrogenation is the main condition for chain initiation, and it requires high energy. This process will reduce the activity of CH4. As for C3H8, its consumption is mainly through H atom extraction by OH, leading to the dominance of isopropyl radicals. C3H8exhibits NTC behavior because the reaction rate of H2O2+(M) = 2OH+(M) at low and high temperatures is faster, resulting in the accelerated reaction rate of OH-related elements. Although the HOCHO chain branching reaction does not produce OH radicals, it consumes active OH and HO2radicals at medium temperatures (800–860 K). The experimental data will help improve the mechanism, and the research results reveal the IDTs of low-carbon hydrocarbons under different conditions at the same time. The experimental data can also provide a theoretical basis for the safe production and conversion of low-carbon hydrocarbons under high temperatures. These results will guide the determination of key parameter safety thresholds and system safety control schemes, which are critical for the safe industrial applications of low-carbon hydrocarbons.
Acknowledgments:This work was supported by the National Natural Science Foundation of China [Grant No. 22278452]. The authors are also thankful to the SINOPEC Research Institute of Safety Engineering for financially supporting this project. Finally, the authors are grateful to the editor and the anonymous reviewers.

- 中国炼油与石油化工的其它文章
- Synthesis and Evaluation of Microporous Metal Organic Frameworks for Light Hydrocarbon Adsorption
- Selection of Extraction Solvents for Bitumen from Indonesian Oil Sands through Solubility Parameters
- Synthesis of Hierarchical Porous Fe2O3/Al2O3 Materials and Study on Catalytic Viscosity Reduction of Heavy Oil
- Activated Carbon from Rice Husk with One-Step KOH Mechanical Mixing Activation as Adsorbent for Treating Phenolic Wastewater
- Effect of Particle Shape on Catalyst Deactivation during 2-Butene and Isobutane Alkylation of Liquid Phase in Fixed-Bed Reactor Using Particle-Resolved CFD Simulation
- Experimental and Numerical Investigation on Erosion Corrosion of the Air Cooler Tube Bundle in a Residue Hydrotreating Unit