
Mechanistic Insights into the Catalytic Cracking of Cyclohexane
2022-02-01 04:02ChenHuiYanJiasongSuYouyouWeiXueerLiRuiWangPengYuShanqingDaiZhenyu
中国炼油与石油化工 2022年4期
Chen Hui; Yan Jiasong; Su Youyou; Wei Xueer; Li Rui; Wang Peng; Yu Shanqing; Dai Zhenyu
(1. College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China; 2. SINOPEC Research Institute of Petroleum Processing Co., Ltd., Beijing 100083, China)
Abstract: Although naphthenes have long been identified as important feedstock components for the production of light olefins and aromatics in fluid catalytic cracking units, their cacking mechanism and microscopic reaction networks, such as activation modes, ring-opening paths, and the production of aromatics, remain debated. In this context, we reported experimental and computational work aimed at elucidating the reaction network of naphthenes in fluid catalytic cracking using cyclohexane as the model naphthene. First, the main reactions for the formation of highly selective and value-added products such as light olefins and aromatics were discussed. Then, the proportions of cyclohexane activation via (i) the non-classical carbonium mechanism and (ii) the classical carbenium mechanism were analyzed by data fitting methods, which revealed that around 32.6% of cyclohexane was initiated by path (i), and the remaining naphthene was activated by path (ii). Moreover, our DFT results showed that the ring opening of cyclohexane through pathway (i) was more difficult than that through path (ii), and ring opening followed by the ring contraction of cyclohexane carbenium ions was the most energetically favorable route among the different ring-opening ways.
Key words: fluid catalytic cracking; naphthene; cyclohexane; alkane activation; ring opening
1 Introduction
Naphthenes are important constituents in fluid catalytic cracking (FCC) feedstocks for producing valuable chemicals such as light olefins and high-octane components[1-2]. The reaction network of naphthenes is very complicated because competing reactions such as cracking, ring opening, ring contraction, hydrogen transfer, and dehydrogenation can occur simultaneously under FCC conditions[3-6], which makes the catalytic cracking mechanism of naphthenes over zeolite catalysts hard to clarify. Although extensive studies regarding the cracking mechanism of naphthenes have been reported[7-11], further research on the reaction network of naphthenes over zeolite catalysts is still needed, especially that focused on the dominant activation modes and ringopening routes, which are still unclear.
In the reaction network of naphthenes, more attention has been paid to ring opening and aromatization reactions because the former is highly desired for the production of value-added products such as light olefins[12-13], and the latter contributes to the formation of aromatics, which are also considered a coke precursor, resulting in the temporary deactivation of the catalyst[14]. Regarding ring opening, two possible mechanisms have been distinguished: direct ring opening and indirect ring opening[15-19]. In the direct ring-opening mechanism, naphthenes are protonated by Brönsted acids or have hydride ion abstracted by hydrogen transfer to acquire the carbonium (activation mode 1) or carbenium ion (activation mode 2), which are further converted into ring-opened products by protolytic cracking or β-scission. In the indirect ring-opening mechanism, naphthenes are activated by hydrogen transfer to give carbenium ions (activation mode 2); the attained carbenium undergoes ring contraction to form five-membered rings which are ultimately transformed into ring-opened products by β-scission[20-21]. The aromatization of naphthenes, which has been reported[22-23], is related to multistep hydrogen transfer reactions, and obtaining cycloalkene from the dehydrogenation of the carbenium ion (activation mode 2) is the key intermediate step for the formation of aromatics[14,24-25]. The above reports have suggested that the catalytic cracking of naphthenes is a multiple-reaction process, and the activation mode of naphthenes plays an important role in the reaction directions. We believe that a deep understanding of the mechanism and reaction network of naphthenes could be helpful to regulate the final product distribution in FCC processes.
In this work, we choose cyclohexane (CHA) as a model naphthene, which has a simple structure and product distribution that facilitate a depiction of the reaction network of naphthenes[25-26]. A combination of product analysis and density functional theory (DFT) is used to study the activation mode, prevalent reaction pathways, and potential ring-opening routes in CHA cracking, providing deep insight into the mechanism of naphthene catalytic cracking. These results will encourage people to explore new catalysts and effective ways to produce value-added chemicals from naphthene-rich feedstocks.
2 Experimental
2.1 Preparation of catalyst
USY Zeolite, kaolin, and silica sol were purchased from Sinopec Catalyst Co., Ltd. Typically, 30% USY zeolite, 40% kaolin, and 30% silica sol were added to a certain amount of deionized water and stirred for 20 min. Then, the slurry was dried and shaped by spray drying technology. After ion exchange with NH4Cl, the solid sample was dried in an oven overnight and calcined in a muffle furnace at 550 °C for 5 h. All obtained catalysts were aged in 100% steam at 800 °C for 8 h before the reaction.
2.2 Characterization of catalyst
The surface area and pore volume of the catalyst were determined on a Micrometrics ASAP 2400 adsorption instrument. Before N2adsorption, the sample was degassed under vacuum at 200 °C for 3 h. The zeolite unit cell size was determined by an X-ray diffractometer (D/MAX 2200PC Rigaku) using Cu Kα radiation (40 kV, 40 mA) with a scanning range of 2θ from 5° to 35° and a scanning rate of 0.01° per second. NH3temperatureprogrammed desorption (NH3-TPD) measurements were conducted on a Micromeritics AutoChemII 2920 instrument. Typically, the adsorption of NH3was performed at 100 °C with 15 mL of NH3/Ar (10 vol% NH3) flow for 1 h. After purging with Ar for 1 h, the temperature was increased from 100 °C to 600 °C at a heating rate of 10 °C/min. The silicon and aluminum content was analyzed by a Panalytical Axois Petro XRF instrument with a rhodium target (50 kV, 50 mA). Table1 shows the main physicochemical properties of the USY zeolite catalyst.

Table 1 Main physicochemical properties of USY zeolite catalyst
2.3 Catalyst evaluation
Fluid catalytic cracking experiments were carried out in an ACE (advanced cracking evaluation) fixed-fluidized bed reactor (built by Kayser Technology, Inc.) at 500 °C. The typical reaction conditions were as follows: the USY catalyst mass was 9 g, the mass ratio of USY/oil was 6, and the weight hourly space velocity (WHSV) was 4 h–1. After each reaction, the reactor was stripped with N2for 450 s. The reaction products were taken from the N2flow at the reactor outlet and analyzed by gas chromatography (Agilent 7890A online for gas products and Agilent 7890B off-line for liquid products) using an FID detector; the selectivity of coke was calculated based on carbon dioxide infrared spectra data collected during the regeneration process of the catalyst.
2.4 Details of the computational model and data fitting method
The density functional theory (DFT) calculations are carried out by Material Studio Dmol3. The generalized gradient-corrected approximation (GGA) and Perdew-Burke-Ernzerhof (PBE) functional with double numeric basis set including p-polarization function (DNP) is used for the geometry optimization calculation. The 19T cluster zeolite model is extracted from H-FAU, as illustrated in Figure 1. The boundary of the zeolite model is saturated with H atoms, forming Si–H dangling bonds. All the Si–H bonds are in the same direction as the zeolite framework, and the Si–H bond lengths are fixed at 0.147 nm. During geometry optimization, only the active region of “SiOHAl(OSi)2OSi” and the reacting molecules are allowed to relax while the rest of the structure is kept fixed at the crystallographic coordinates[22].
The calculated product distribution given in section 3.3 was obtained by data fitting between CHA and “substitute intermediates” (hexene and cyclohexene represent cyclohexane carbonium and cyclohexane carbenium intermediates, respectively) cracking products. The fitting formula was as follows:
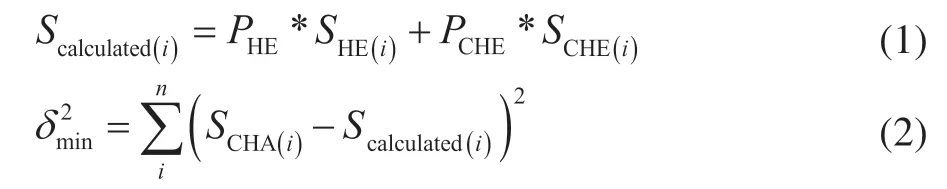
The percentages of cyclohexane cracked through hexene and cyclohexene intermediates are denoted asPHEandPCHE, respectively.SHE(i)andSCHE(i)represent the selectivity of the hexene and cyclohexene cracking products, respectively. The iteration method is used for computing the minimum variance, which makes the selectivity of the calculated product distribution best fit the CHA cracking product distribution.
3 Results and Discussion
3.1 Product distribution
The cracking of CHA on the USY catalyst led to a mixture of products containing more than 50 components. The main products obtained are shown in Figure 2. The reaction products included H2, C1–C6linear hydrocarbons, cycloalkanes, cycloalkenes, aromatics, and coke. To facilitate the identification of the main reactions in CHA conversion, the products were classified into the following groups: (1) isomerization products, with a selectivity of 45.7%, containing five-membered rings (i.e., methylcyclopentane and methylcyclopentene). Their formation could be explained by the ring contraction of CHA[20]; (2) dehydrogenation products, including unsaturated six-membered rings (e.g., cyclohexene, benzene, and toluene), generated from hydrogen transfer and direct dehydrogenation[26]. The selectivity of the dehydrogenation products was 28.9%; (3) cracking products, with a selectivity of 25.4%, including H2, C1–C6linear paraffin, and olefins formed via protolytic dehydrogenation, ring opening, and cracking[14].
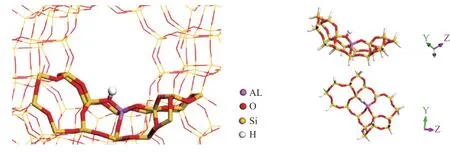
Figure 1 19T cluster model of H-FAU zeolite
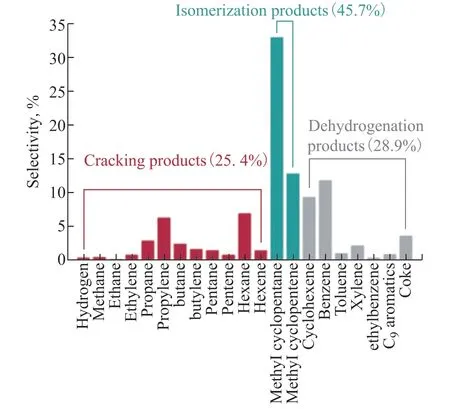
Figure 2 Product selectivity for cyclohexane (CHA) cracking on USY catalyst
According to the product distribution, the catalytic cracking of CHA on the USY zeolite catalyst is a complicated process involving various products and reactions. The prevailing reactions for the highly selective products here are ring contraction, hydrogen transfer, dehydrogenation, ring opening, and cracking.
3.2 Reaction pathways and reaction network
At low conversion rates (generally <10%), the products of CHA conversion could be considered generated by primary reaction (accompanied by negligible secondary reaction). Thus, the reaction pathways were feasibly illustrated based on product analysis. The main reaction pathways for the formation of the highly selective products are discussed as follows.
H2could be produced by either the direct dehydrogenation or protolytic dehydrogenation of CHA simultaneously[14,19], whereas the direct dehydrogenation of naphthene generally occurred on a catalyst with metal sites or under harsh reaction conditions[27-28]. Considering the absence of metal sites and the high energy barrier of dehydrogenation under FCC conditions[29], H2should be given by the protolytic dehydrogenation of CHA (Scheme 1a).
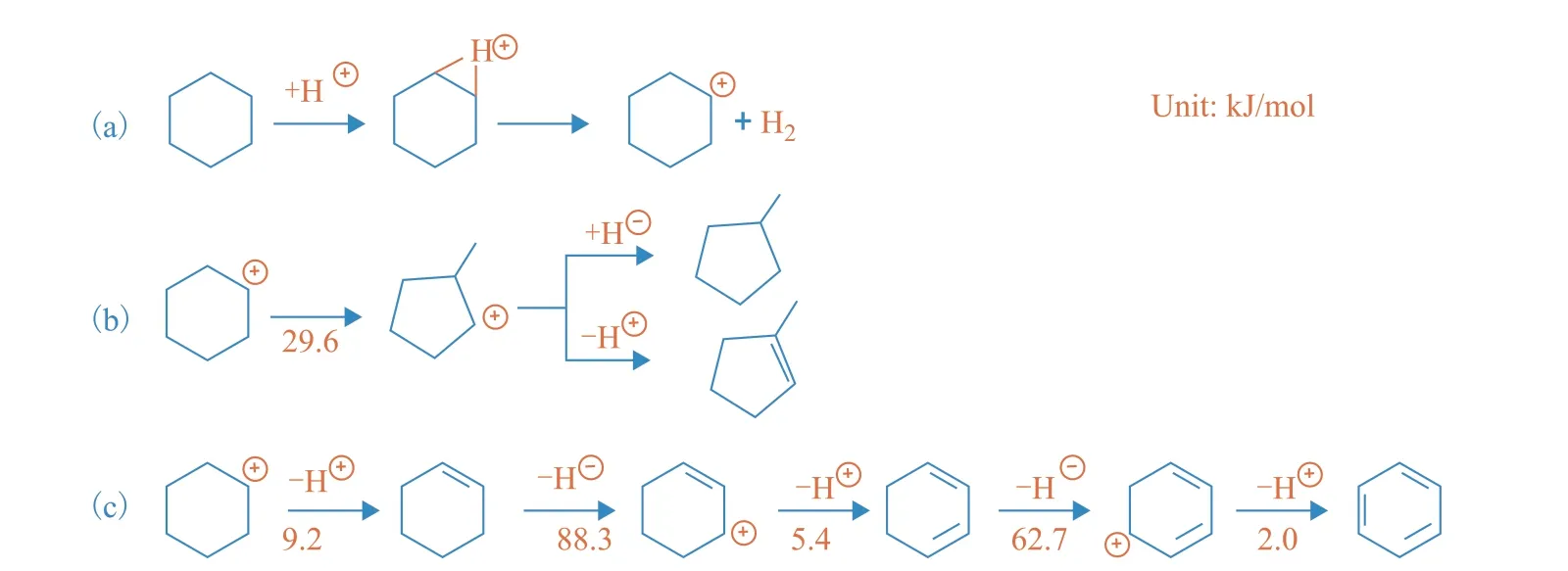
Scheme 1 Reaction paths of (a) protolytic cracking, (b) ring contraction, and (c) hydrogen transfer
The isomerization products showed the highest selectivity among the three groups of products. The CHA carbenium ion generated from protolytic dehydrogenation (Scheme 1a) underwent ring contraction to yield the methylcyclopentane carbenium ion, which gave H+to the catalyst surface or abstracted H−from hydrogen donors, ultimately producing isomerization products (Scheme 1b). The high selectivity of the isomerization products also implied that ring contraction was an easy step to perform under the reaction conditions. A low energy barrier for the ring contraction of CHA (29.6 kJ/mol) was consistent with the experimental observations. To further prove this point, we compared the product distribution of cyclohexene and methylcyclopentene (Because cyclohexene and methylcyclopentene could be protonated easily at the C=C bond and lead to CHA and methylcyclopentene carbenium ions, respectively, it was reasonable to use related cycloalkenes to represent the cracking of CHA and methylcyclopentene carbenium ions). As shown in Figure 3, cyclohexene and methylcyclopentene had similar product distributions, which confirmed that the ring contraction process was apt to happen.
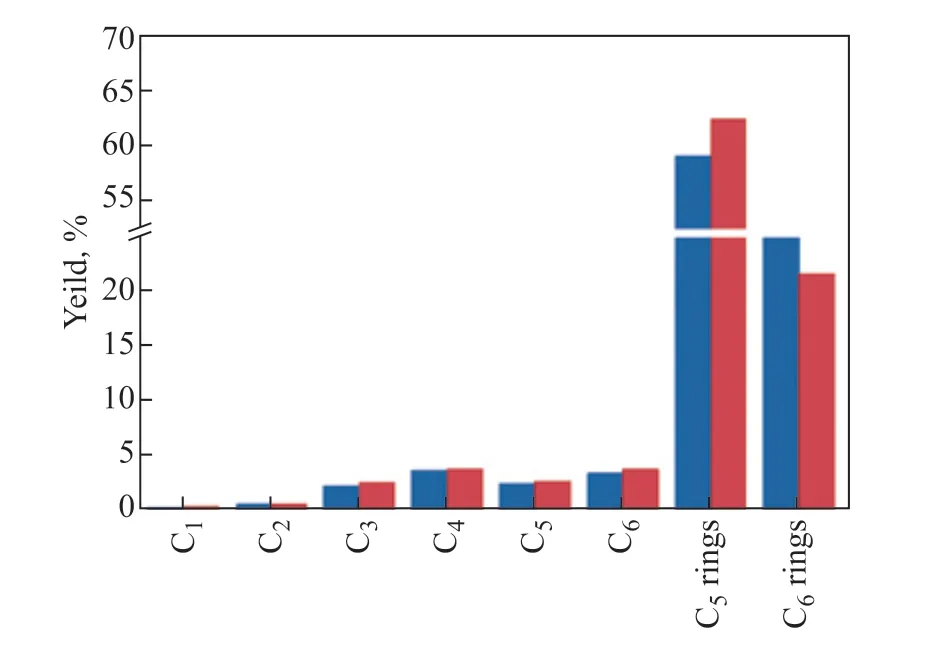
Figure 3 Comparison of product yields of cyclohexene and methylcyclopentene
The formation of dehydrogenation products involved hydrogen transfer and dehydrogenation reactions. Considering it was difficult for dehydrogenation to occur under the reaction conditions (as mentioned regarding the formation of H2), hydrogen transfer should be the main reaction accounting for the dehydrogenation products. CHA was firstly activated by hydrogen transfer to yield the CHA carbenium ion, which continued to lose hydrogen through five consecutive bimolecular hydrogen transfer reactions (two steps of hydride transfer and three steps of proton transfer), giving cyclohexadiene and finally benzene (Scheme 1c)[30]. According to the DFT results, the hydride transfer steps had higher energy barriers (88.3 kJ/mol and 62.7 kJ/mol for cyclohexene and cyclohexadiene hydride transfer, respectively) compared to proton transfer (9.2 kJ/mol, 5.4 kJ/mol, and 2.0 kJ/mol for CHA carbenium ion, cyclohexene carbenium ion, and cyclohexadiene carbenium ion proton transfer, respectively), which suggested that cyclohexene hydride transfer leading to the cyclohexene carbenium ion was the rate-limiting step in the conversion process of CHA to benzene (except the activation step of CHA). Moreover, we noted that cyclohexadiene was an unstable product, which was apt to undergo protonation reaction (17.6 kJ/mol) or hydride transfer (62.7 kJ/mol). Therefore, none of it was identified in the GC product analysis.
Cracking species was another highly selective product in CHA conversion. Three possible ring-opening pathways were responsible for cracking products: (i) ring-opening path I, as shown in Scheme 2a, where gas-phase CHA is initiated at the Brönsted acid site on the USY catalyst and forms a CHA carbonium ion by protonation. The activated species undergoes protolytic cracking to give a hexane carbenium ion and ultimately transform into C1–C6alkanes and alkenes through isomerization, cracking, and hydrogen transfer; (ii) ring-opening path II, as shown in Scheme 2b, where an activated CHA carbenium ion (produced by hydride transfer from CHA to catalyst surface) undergoes ring opening to obtain a hexene carbenium ion which is subsequently converted into C1–C6alkanes and alkenes; and (iii) ring-opening path III, as shown in Scheme 2c, where the methylcylopentane carbenium ion (produced by the ring contraction of a CHA carbenium ion), as an important intermediate, undergoes ring opening to generate the hexene carbenium ion, which subsequently yields C1–C6alkanes and alkenes via isomerization, cracking, and hydrogen transfer reactions.

Scheme 2 Reaction paths of (a) ring-opening path I, (b) ring-opening path II, and (c) ring-opening path III
Based on the above analysis, the reaction network for CHA cracking was established (Figure 4). During the cracking process, different reaction paths took place simultaneously. All these reaction paths consisted of the reaction networks of CHA and together determined the final product distribution.
3.3 The proportion of two activation modes
As shown in Figure 4, the cracking behavior of CHA was related to the active intermediates (i.e., the CHA carbonium ion and the carbenium ion). For example, if CHA cracked via the carbonium ion intermediate, then protolytic cracking and ring opening would be the main reactions accompanied by the increasing selectivity of H2and C1–C6alkanes and alkenes. In contrast, if CHA was cracked through a carbenium ion intermediate, the selectivity of five-membered rings and six-membered rings may increase. Thus, by clarifying the proportion of two kinds of active intermediates in CHA cracking, the contributions of the different activation modes to the product distribution could be determined.
As mentioned before, cyclohexene could be protonated easily at the C=C bond of the acid site and lead to the CHA carbenium ion. Thus, it was reasonable to use cyclohexene as a substitute for the CHA carbenium ion as an intermediate and simulate the cracking of the CHA carbenium ion, whereas it seemed difficult to find a stable compound representing the CHA carbonium ion, which has a five-coordinated structure. Here, we found the CHA carbonium ion was apt to cleave C–C (0.2 kJ/mol) and produce hexane carbenium ion species, which were easily given by the protonation of hexene at the Brönsted acid site. Therefore, hexene could be an appropriate model compound to represent the CHA carbonium ion. Cracking tests of the “substitute intermediates” (i.e., cyclohexene and hexene) were performed on the USY catalyst under the same condition for CHA cracking. The product distribution of “substitute intermediates” was shown in Table 2. Figure 5 gives a comparison of the experimental and calculated CHA product distribution (The calculated result came from cyclohexene and hexene cracking results treated by data fitting methods. Details are given in section 2.4).
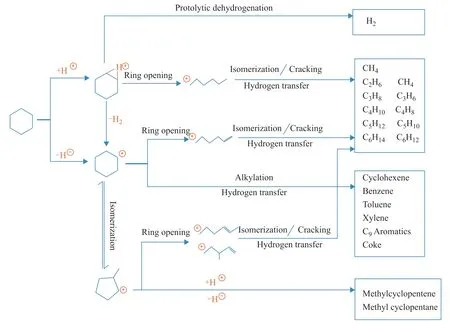
Figure 4 Reaction network for catalytic cracking of cyclohexane

Table 2 Products slate of different feedstocks over USY catalyst
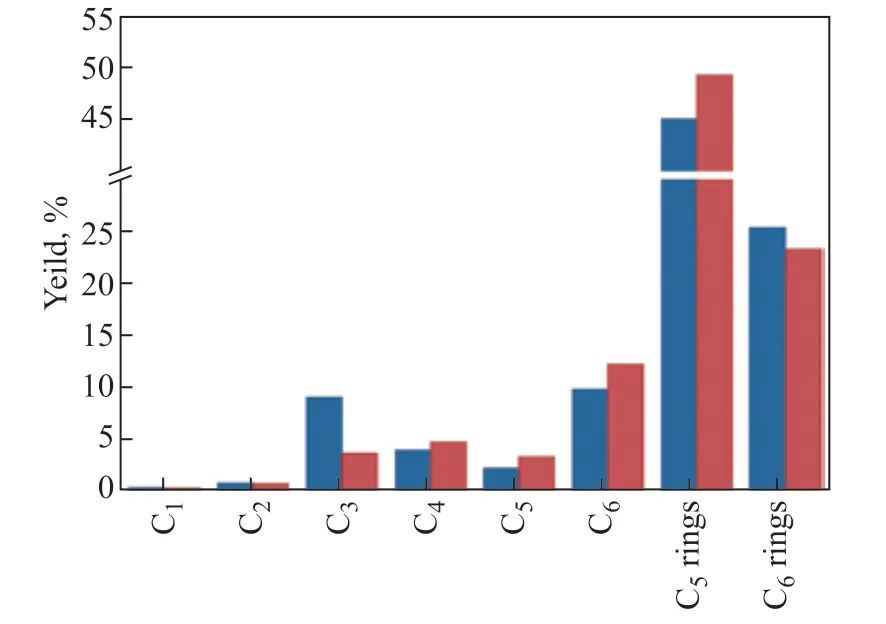
Figure 5 Comparison of products yield of cyclohexane cracking and calculated results ■—Cyclohexene cracking; ■—Calculated
In Figure 5, except for the calculated yield of C3being significantly lower than that of the experimental results, there was no significant difference between the experimental and calculated product distributions. The lower calculated C3yield could be related to the high conversion of cyclohexene and hexene, as high conversion in FCC usually results in C3oligomerization, which led to the decrease of C3yield and the increase of six-membered and five-membered ring yields[23]. Because the “substitute intermediates” simulated the cracking of CHA very well, we obtained the proportions of carbonium ion and carbenium ion intermediates under typical experimental conditions, which were 17.6% and 82.4%, respectively. Considering that CHA carbenium ions may be partially derived from the protolytic dehydrogenation of CHA carbonium ions, as mentioned in section 3.1, and the quantity of this transformation was equal to the yield of H2(Scheme 1a), the real proportions of carbonium ion and carbenium ion intermediates in CHA cracking should be 32.6% (17.6% + 15.0%) and 67.4% (82.4% − 15.0%), respectively. Therefore, the amount of CHA activated via the carbonium ion was less than that of the carbenium ion.
3.4 Energy for different ring-opening pathways
The ring opening of naphthene is an important process for value-added chemical products such as light olefins, so a deep understanding of the ring-opening mechanism is very meaningful. According to Scheme 2, different ring-opening paths had a similar product distribution. Thus, it was difficult to judge which ring-opening path was the dominant one when only considering the product analysis. Therefore, we attempted to address the issue by comparing reaction energy calculations[31-35].
In the ring-opening path I (Figure. 6), the proton on the Brönsted acid site is inserted into the C–C bond of CHA and leads to a carbonium ion, which subsequently cracks the C–C bond to form an adsorbed hexane carbenium ion. The energy barriers of protonation and C–C bond cleavage are 176.0 kJ/mol and 0.2 kJ/mol, respectively, which are close to the estimated energies of protolytic cracking reported by Maihom[35].In ring-opening path II (Figure 7), the energy barrier of hydride transfer from CHA to the adsorbed C3carbenium ion is 110.7 kJ/mol. The resulting CHA carbenium ion subsequently surmounts a β-scission energy barrier of 45.1 kJ/mol to give a 1-hexene carbenium ion.

Figure 6 Energy profile for the mechanism of ring-opening path I
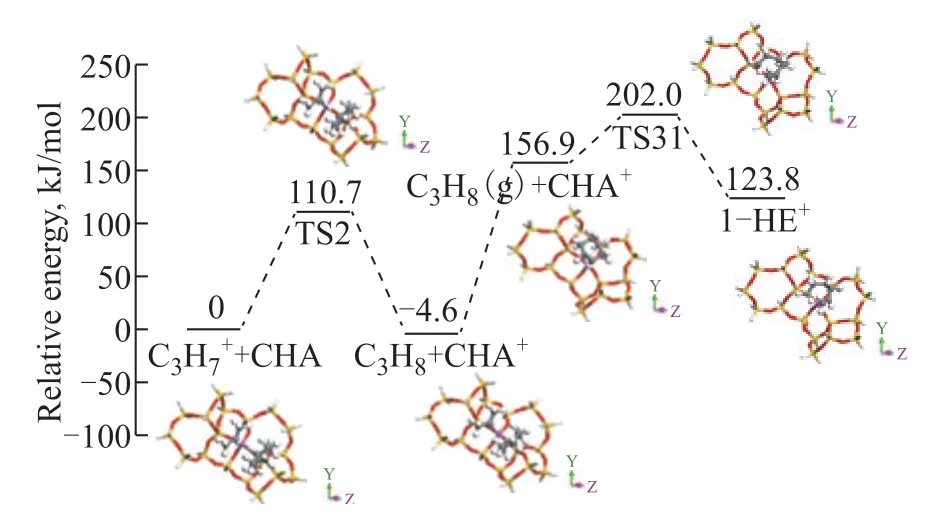
Figure 7 Energy profile for the mechanism of ring-opening path II
In ring-opening path Ⅲ (Figure 8), the energy barrier of CHA carbenium ion ring contraction to form the MCPA carbenium ion through the protonated cyclopropane transition state is 29.6 kJ/mol. The β-scission of the methylcyclopentane carbenium ion forms an absorbed 2-hexene carbenium ion, with a barrier of 12.1 kJ/mol.
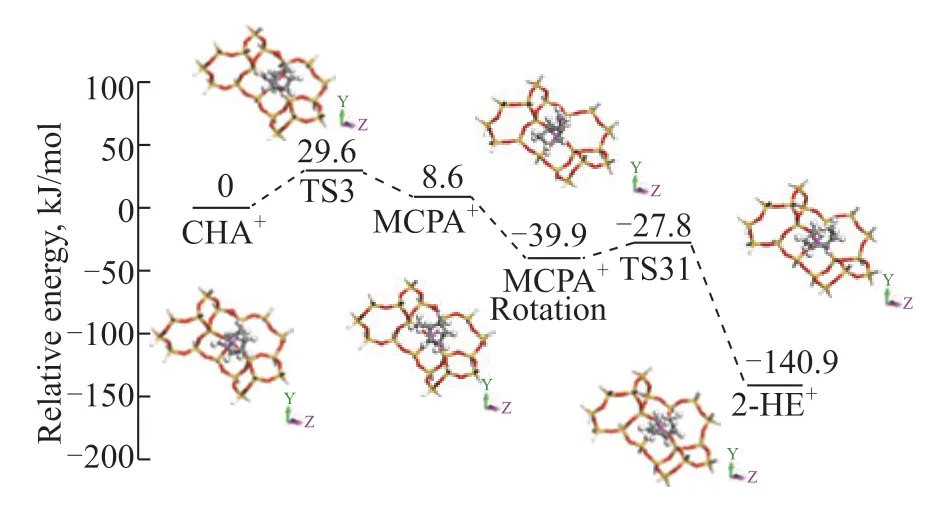
Figure 8 Energy profile for the mechanism of ring-opening path III
The calculation results implied that the activation of CHA via carbonium was more difficult than that via the carbenium intermediates, which means it was hard for the ring-opening path I to take place compared to other ring-opening routes in CHA conversion. Nevertheless, it is worth noting that once the CHA carbonium ion was formed, it was easy for ring opening to occur due to the extremely low reaction barrier. Additionally, the energy barrier of ring-opening path III was found to be lower than that of ring-opening path II, even though both reaction paths had the same activated intermediate (the CHA carbenium ion). The three ring-opening paths follow the energy order of ring-opening path I > ring-opening path II > ring-opening path III. Therefore, ring-opening path III was the most energy-favored reaction path in the CHA cracking process.
4 Conclusion
The reaction network of cyclohexane was systematically investigated, and 25.4%, 45.7%, and 28.9% of cyclohexane was converted into cracking, isomerization, and dehydrogenation products, respectively. Carbonium and carbenium ions were important intermediates in the cracking of model naphthenes, and they determined the reaction direction of cyclohexane. In the activation step, around 32.6% of cyclohexane was converted into carbonium ions by crossing a high energy barrier of 176 kJ/mol. The obtained carbonium ions were prone to cleave the C–C bond to produce C1–C6linear products (ring-opening path I). However, when cyclohexane was converted into carbenium ions with an energy barrier of 110.7 kJ/mol, the formed carbenium ion was apt to undergo ring contraction and 5-memebered ring opening (ring-opening path III) rather than directly cracking the 6-membered ring (ring-opening path II). Finally, it is important to note that the carbenium ion surmounted an 88.3 kJ/mol energy barrier and could transform into aromatics, but this process should be avoided when a ring-opened compound is the target product.
Acknowledgment:This work was performed with the financial support of Sinopec Research Institute of Petroleum Processing (RIPP, Proj. R17022).

- 中国炼油与石油化工的其它文章
- Synthesis and Evaluation of Microporous Metal Organic Frameworks for Light Hydrocarbon Adsorption
- Selection of Extraction Solvents for Bitumen from Indonesian Oil Sands through Solubility Parameters
- Synthesis of Hierarchical Porous Fe2O3/Al2O3 Materials and Study on Catalytic Viscosity Reduction of Heavy Oil
- Activated Carbon from Rice Husk with One-Step KOH Mechanical Mixing Activation as Adsorbent for Treating Phenolic Wastewater
- Effect of Particle Shape on Catalyst Deactivation during 2-Butene and Isobutane Alkylation of Liquid Phase in Fixed-Bed Reactor Using Particle-Resolved CFD Simulation
- Experimental and Numerical Investigation on Erosion Corrosion of the Air Cooler Tube Bundle in a Residue Hydrotreating Unit