
Transcriptome Analysis of the Digestive Tract of Tachypleus tridentatus and Carcinoscorpius rotundicauda
2022-06-14 06:54LIYuhongKWANKitYueLISansuLIUYingchunWENGBosenHUANGWenshuandWENGZhaohong
LI Yuhong, KWAN Kit Yue, LI Sansu, LIU Yingchun, WENG Bosen,HUANG Wenshu, and WENG Zhaohong
Transcriptome Analysis of the Digestive Tract ofand
LI Yuhong1), *, KWAN Kit Yue2), LI Sansu3), LIU Yingchun4), WENG Bosen1),HUANG Wenshu5), and WENG Zhaohong5)
1),,361021,2),,535011,3),,361021,4),,350108,5),,361021,
Transcriptome assemblies for the stomach, midgut, and fecal mucosa ofandare reported for the first time in this study. Genome assembly resulted in 373069 unigenes with an N50 of 1314, while transcriptome assembly resulted in 736378 unigenes and an N50 of 2121. The annotated unigenes showed the highest similarity to fishes, amphibians, and reptiles, with most unigenes closely related to metabolism, translation, biogenesis, signal transduction, en- ergy production, immune response, and secretion. Unigenes (4768) were mapped to KEGG pathways, and the most enriched path- ways were involved in translation, environmental information signal processing, metabolism, endocrine system, immune system, ner- vous system, and varying metabolism. Totally 65889, 2001, and 7162 differentially expressed genes (DEGs) were identified in the stomach, midgut, and fecal mucosa between the two horseshoe crab species, respectively. Compared with, 99.95% of the DEGs in thestomach were down-regulated, while 99.97% of the DEGs offecal mucosa and 55.42% of the DEGs of themidgut were up-regulated. Most midgut DEGs were involved in hydrolase activity, protein metabo- lism, and cell cytoplasm, while most stomach and fecal mucosa DEGs were involved in catalytic activity and primary metabolic processes. Most stomach DEGs were assigned to the cellular component in cellular macromolecular complexes, while most fecal mucosa DEGs were assigned to the cellular component in cell and cell parts. These results will benefit the investigation of the mo- lecular mechanisms of the digestive tract related to the feeding habits and environmental traces of horseshoe crabs.
transcriptome; digestive tract;;; differentially expressed genes (DEGs)
1 Introduction
The horseshoe crabs, as well-known classic ‘living fos- sils’, have survived for nearly 500 million years (Van, 2010). Their survival is inseparable from their feeding ha- bits, morphological structure, and physiological and meta- bolic characteristics, including omnivory, strong immunity,and tolerance to environmental changes.,,,andare the four species of horseshoe crabsin the world (Wang, 2020).andhave been captured since the 1980s to providepharmaceutical manufacturers with/amebocyte lysate (LAL/TAL) by using their blood as the raw material (Novitsky, 2015). In the modern medical sys- tem, every injection, every intravenous infusion, and every medical device that needs to be implanted into the human body need the LAL/TALreagent for endotoxin testing.andare listed as Vulnerable (VU) (Smith, 2016) and Endangered (EN) species (Laurie, 2019),respectively, on the International Union for Conservation of Nature (IUCN) Red List. High tide spawn- ing sites and intertidal juvenile nursery habitats are criti- cal to the horseshoe crab’s survival (Laurie, 2019). The overharvesting and habitat loss with the development of coastal economic zones have severely affected the sur- vival of horseshoe crabs. All four horseshoe crab species worldwide are now endangered (Wang, 2020). Bothanddistributed in China have been strictly protected by law as National Second-class Pro- tected Wildlife Species since February 2021. Recently, thesignificant population declines have gained in notoriety due to the integration of science, management, education, and policy for strengthening conservation research. Habitat pro-tection has been considered one of the best strategies to conserve horseshoe crabs (Chen., 2015; Laurie, 2019).
All living animals depend on food for their survival. Feed- ing habits, digestion, and absorption are directly related to important life activities such as growth and reproduction. The horseshoe crabs can change their nutritional sources and upgrade their position in the food chaingrowth (Gaines, 2002). The digestive tract of horseshoe crabis partially formed at hatching and becomes functional be- fore feeding begins. The digestive tract plays a vital roleduring the digestion in the animals. The structure and func- tion of the digestive tract of the horseshoe crab are close- ly related to its omnivorous feeding habit (Hong, 2011). The external morphology and anatomy of the digestive tracts ofandare similar (Chen, 2016). The basic internal gross anatomy of horse-shoe crabs has been described. The digestive tract is di- vided into foregut, midgut, and hindgut three parts (Chat- terji., 1988; Hong, 2011). The foregut includes the cavum buccale, esophagus, stomach/proventriculus, and py- lorus. The hindgut consists of the rectum and anus (Chen,, 2016). There is no remarkable difference in structure between midgut and hindgut (Xie, 2018).
At present, many research reports have been published concerning the feeding habits and digestive metabolism of horseshoe crabs, generally focusing on their food sources and the anatomy of the digestive tract (Gaines,, 2002;Carmichael., 2009; Hong, 2011; Chen,, 2016;Fan., 2017). However, the mechanisms of feeding,digestion, and absorption in horseshoe crabs remain unclear, and information on the transcriptome of digestive organs forandis limited. The trans-criptome evolution of different species can be comprehend- ed by collecting and comparing transcriptome data (Wang, 2021). In this study, high-throughput Illumina So- lexa sequencing and gene annotation were used to char- acterize the transcriptome of the digestive tract and mass mucosa related to the digestive metabolism ofand. The first comparative analysis of large-scale gene expression profiles betweenandjuveniles is reported hereinThus, the transcriptome analysis reported in this article is helpful for understanding the molecular mechanisms of the adaptive evolution related to the digestive metabolism ofand
2 Methods
2.1 Horseshoe Crabs and Sample Preparation
Experimental animal samples were collected in accor- dance with the ethical principles of animal experimentation.The materials were stomach (S), midgut (G), and fecal mu- cosa (F) isolated from three individuals ofand, respectivelyThe animals were all healthy juveniles with 3.3cm±0.5cm of prosomal width. The animals were obtained from the intertidal zone in the west bay of northern Beibu Gulf, Guangxi, China at the end of March 2019. The juveniles were transported to the laboratory and acclimated to laboratory conditions for three weeks. All horseshoe crab juveniles were cultured in aqua- rium tanks (dimensions: 120cm×40cm×25cm) equipped with a water filtration system, thermostatic heaters, and ul- traviolet sterilizers. A 4-cm sediment layer was provided underneath. Seawater was maintained under the following conditions: temperature 26–30℃, salinity 32–33, pH 7.6–7.9, dissolved oxygen 6–7mgL−1, and a light:dark pho- toperiod of 12h:12h. During the acclimation period, the horseshoe crab juveniles were fed a daily ration of 2% of their biomass with frozen brine shrimp and clam meat. The juveniles were randomly transferred from the acclimation tank to the aquaria used for the experiments and kept for two days without food until the beginning of the experi- ment. Three tissue samples from three horseshoe crab ju- veniles were collected, and each tissue sample was cut into small pieces and put into 2-mL RNase-free centrifuge tube. An appropriate volume of RNA Later (RNA Stabilization Reagent, QIAGEN) was added, and the sample was stored at −80℃. For easy identification of the samples, three sto- mach samples ofwere named TTS1, TTS2, and TTS3; three midgut samples ofwere named TTG1, TTG2, and TTG3, and three fecal mucosa samples ofwere named TTF1, TTF2, and TTF3. The three stomach samples ofwerenamed CRS1, CRS2, and CRS3; the three midgut samples ofwere named CRG1, CRG2, and CRG3; and the three fecal mucosa samples ofwere named CRF1, CRF2, and CRF3.
2.2 RNA Extraction and Quality Control
Total RNA was extracted from the 18 tissue samples withthe RNeasy MiNi Kit (QIAGEN, USA) in accordance with the manufacturer’s instructions. The quality of RNA sam- ples was controled with several aspects. First, RNA degra- dation and possible contamination were monitored by 1% agarose gel electrophoresis. Then, the purity, concentration, and integrity of total RNA were respectively measured us- ing the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA), the Qubit ® RNA Assay Kit in Qubit ® 2.0 Fluorometer (Life Technologies, CA, USA), and the RNANano 6000 Assay Kit of the Bioanalyzer 2100 system (Agi- lent Technologies, CA, USA).
2.3 Library Preparation for Transcriptome Sequencing
Library preparation for transcriptome sequencing was performed by conventional methods of molecular biology. RNA sequencing samples were prepared based on 1.5µg RNA per sample. Sequencing libraries were constructed using a NEBNext®Ultra™ RNA Library Prep Kit for Illu- mina®(NEB, USA) according to the manufacturer’s in- structions. Briefly, oligo (dT)-attached magnetic beads were used to purify mRNA from total RNA. Library quality was assessed with the Agilent Bioanalyzer 2100 system. Li- braries were sequenced by the Illumina Hiseq platform fol-lowing the manufacturer’s protocols, and paired-end reads were obtained.
2.4 Transcriptome Assembly
High-quality clean reads obtained by removing reads containing poly-N and reads containing Illumina adapters, and low quality reads from raw data were subjected to sub-sequent analysis based on the methods of quality control(Zhang, 2017). In the present study, at least 6Gb clean data were obtained for each sample. Trinity’s utility (Grabherr. 2011) was used to performtran- scriptome assemblies.
2.5 Data Analysis
2.5.1 Comprehensive gene annotation
We performed gene functional annotation using the fol- lowing databases. The NCBI nonredundant protein se- quences (NR) database was used for identifying putative mRNA functions. The Protein family (Pfam) database was used for classifying protein sequences into families and do-mains. The possible functional classifications and mole- cular pathways were predicted with databases including NCBI nonredundant nucleotide sequences (NT), Clusters of Orthologous Groups of proteins (KOG/COG), a high- quality manually annotated and reviewed protein sequencedatabase (SWISS-PROT), the KEGG Ortholog database (KO), and Gene Ontology (GO).
2.5.2 Differential expression analysis
For the samples in the present experiments, differential gene expression analysis of two experimental groups was conducted using the DESeq R package (1.10.1). The Ben- jamini and Hochberg approach for controlling the false dis-covery rate was applied to adjust the resultingvalues.Genes with adjusted-values below 0.05 were determined to be differentially expressed. GO enrichment analysis of the differentially expressed genes (DEGs) was conducted by the GOseq R packages (Young, 2010).
3 Results
3.1 Summary of Sequencing Data
High-quality sequencing and mapping results were ob-tained. The summary statistics of the sequencing data are shown in Table 1. After removing the low-quality reads, we obtained clean reads ranging from 40.37 million to 52.95 million, and clean bases ranging from 6.06Gb to 7.94Gb from the stomach, midgut, and fecal mucosa samples ofThe number of clean reads from three ex- perimental tissues ofranged from 43.24 mil-lion to 56.13 million. Correspondingly, the number of clean bases ranged from 6.49Gb to 8.42Gb (Table 1). The av- erage GC content of the transcriptome data ofstomach (TTS) was 37.36%, a value slightly lower than that ofwhich was 40.74%. The av- erage GC contents of the transcriptome data ofmidgut (TTG) and fecal mucosa (TTF) were 44.29% and 40.71%, respectively, slightly higher than those of, which were 40.08% and 38.81% (Table 1). Clean reads (73.62%–79.92%) from 18 samples could be mapped to the reference genome, suggesting that the qua- lity of our sequencing data was high. The genome assem- bly resulted in 373069 unigenes with an N50 of 1314, whilethe transcriptome assembly resulted in 736378 unigenes and an N50 of 2121. These 373069 unigenes could be assign- ed to the protein database for annotation information. Next, we analyzed the length distributions of the unigenes and transcripts in the assembly samples (Fig.1). The N50 va- lue of transcript length was longer than 1100bp, and the N50 value of unigene length was longer than 900bp.
3.2 Functional Annotation and Classification of the Assembled Unigenes
For the purpose of predicting the functions of the uni- genes, all 373069 sequences were analyzed by the data- base, and the success rates of the annotated data are listed in Table 2. Among the 373069 unigenes, more than 30% showed significant matches in the PFAM database, GO database, NT database, NR database, or Swiss-Prot data- base. A total of 132594 unigenes (35.54%) showed sig- nificant matches in both the PFAM and GO databases.

Table 1 Summary of sequencing data of stomach (S), midgut (G), and fecal mucosa (F) of T. Tridentatus and C. rotundicauda
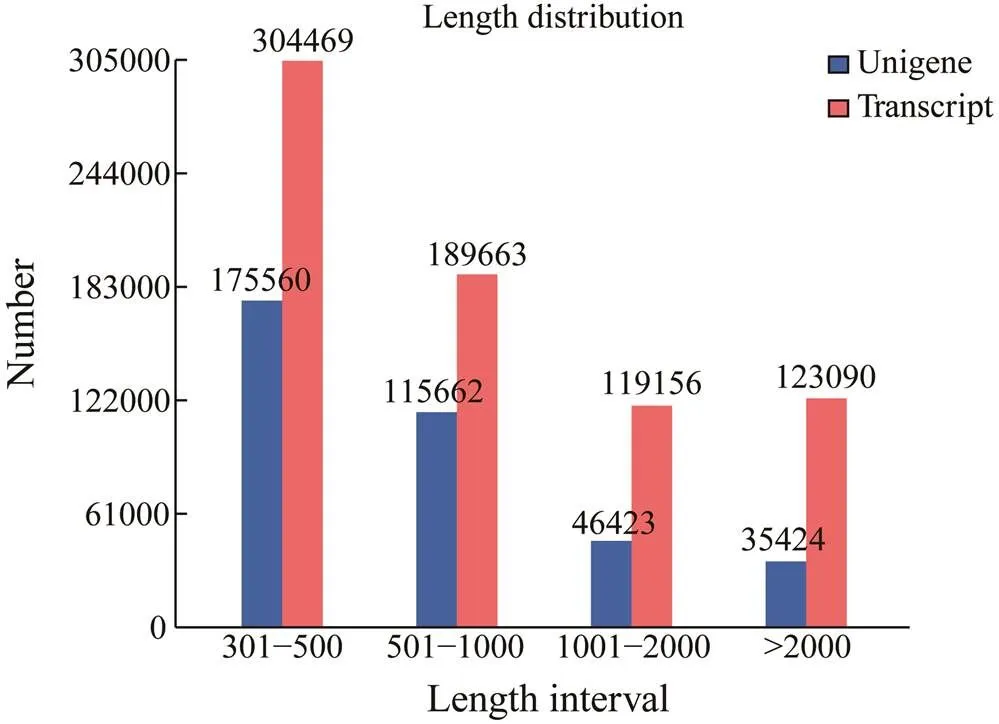
Fig.1 Length and quantity distributions of transcripts and unigenes.

Table 2 Gene annotation success rate statistics
Notes: Annotated in at least one database: the number and percent- age of unigenes successfully annotated with at least one database.
There were 10482 genes in the digestive tracts of thehorseshoe crabs that exhibited significant matches with those of. The top 10 species among these matched unigenes were,,,,,,,,,andThe ratio of the species classification with the highest similarity to the annotated genes is shown in a pie chart (Fig.2). These species were distributed among fishes, amphibians, and reptiles. This result may provide clues for the biological evolution of horseshoe crabs.

Fig.2 Species classification of genes in the transcriptome.
The GO annotations of the assembled unigenes from the transcriptomes ofanddiges- tive tract are shown in Fig.3. There were 132594 contigs assigned to three GO terms classified as cellular component (CC), biological process (BP), and molecular function (MF). These were assigned to 26, 20, and 10 subcategories, res- pectively. For biological process, cellular process (76617, 50%), including cellular macromolecule metabolic process(11%) and protein metabolic process (5.5%) were the most represented, followed by metabolic process (70900, 45.3%), single-organism process (62048, 39.7%), biological regu- lation (28216, 18%), regulation of biological process (26597, 17%), and response to stimulus (18746, 5.2%). Under the molecular function category, binding (64517, 41.3%) in- cluding nucleic acid binding (13.1%) was the most abun- dant, followed by catalytic activity (59773, 38.2%) and transporter activity (10947, 7%). Among the cellular com- ponent terms, cell (37831, 22.1%) and cell part (37831, 22.1%) occupied the same dominant subcategory (Fig.3). These annotations represent a profile for gene expression ofand,suggesting that there were diverse protein coding genes in the digestive tract closely related to digestion, transportation, and nutrient ab- sorption. These results offer many research resources for further investigation of cellular, metabolic, regulatory, bind- ing, catalysis, and transport mechanisms. In addition to a large number of the annotated genes being assigned to di- gestion, absorption, and transportation processes, some im- portant genes involved in innate immune systems andner- vous systems were also found.

Fig.3 GO annotations of transcriptomic data of T. tridentatus and C. rotundicauda.

Fig.4 KOG annotations of transcriptomic data in T. tridentatus and C. rotundicauda.
Among 373069 assembled unigenes, a total of 4768 uni- genes were mapped to KEGG pathways (Fig.5) and cate- gorized into 32 subclasses. The most enriched pathways were translation (599 members), environmental information signal processing/transduction (554 members), amino acidmetabolism (503 members), carbohydrate metabolism (488members), endocrine system (393 members), folding, sort- ing and degradation (285 members), immune system (237 members), nervous system (197 members), nucleotide me- tabolism (189 members), digestive system (180 members), metabolism of cofactors and vitamins (179 members), andenergy metabolism (167 members). This demonstrated thatour transcriptome database had various unigenes related to metabolism and environmental information signal process-ing/transduction. A well-categorized and annotated resourceof the transcriptome in digestive tract ofandcan become a valuable database for inves- tigating their specific bioprocesses and identifying impor- tant functional genes related to the physiological process- es of digestion and absorption.
3.3 Differentially Expressed Genes Between T. tridentatus and C. rotundicauda
The stomach (TTS, CRS), midgut (TTG, CRG), and fe- cal mucosa (TTF, CRF) transcriptomes ofandwere analyzed for the presence of DEGs in comparisons of the two horseshoe crab species.The total numbers of up- and down-regulated DEGs in each tissue are listed in Table 3. Venn diagrams depictthe over- all distribution of DEGs in each of the comparisons. Dif- ferential expression analysis revealed that there were 65889 DEGs between thestomach andstomach, while there were 2001 unigenes differen- tially expressed betweenmidgut andmidgut; 1109 unigenes were up-regulated, and 892 were down-regulated. In a total of 7162DEGs in the comparison betweenfecal mucosaandfecal mucosa,7160 unigenes were up-regu- lated, and only two genes were down-regulated. Venn dia- grams (Fig.6) depict the overlap between different sets of DEGs in the three comparisons of three tissues in the two species. Compared with, 99.95% of the DEGs in thestomach were down-regulated, while 99.97% of the DEGs offecal mucosa and 55.42% of the DEGs of themidgut were up-regulated.

Table 3 Numbers of differentially expressed genes between T. tridentatus and C. rotundicauda
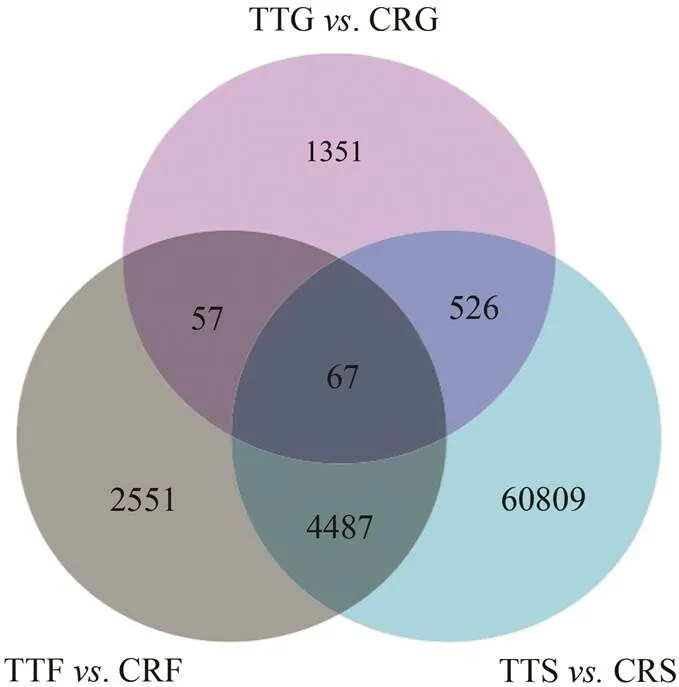
Fig.6 Venn diagram ofdifferential gene expression analysis between T. tridentatus and C. rotundicauda.Threshold values FDR<0.05 are used to control false discovery rates, all differentially expressed genes (DEGs) are at least 2-fold up- or down-regulated.
3.4 GO Functional Classification of DEGs
After identifying the DEGs, the top 20 terms of the GO enrichment analyses in each basic functional category were selected, and their basic functions combined with GO an- notation were described. A good understanding of the bio- logical function of the digestive tract groups betweenstomach (TTS) andstomach (CRS)could be obtained by the standard GO classification.The DEGs of GO functional classification were assigned into three functional groups: biological process, molecular function, and cellular component. Three categories of GO functional classification comparing the tissue groups (TTS– CRS, TTG–CRG, and TTF–CRF) are shown in Figs.7A, B, C.
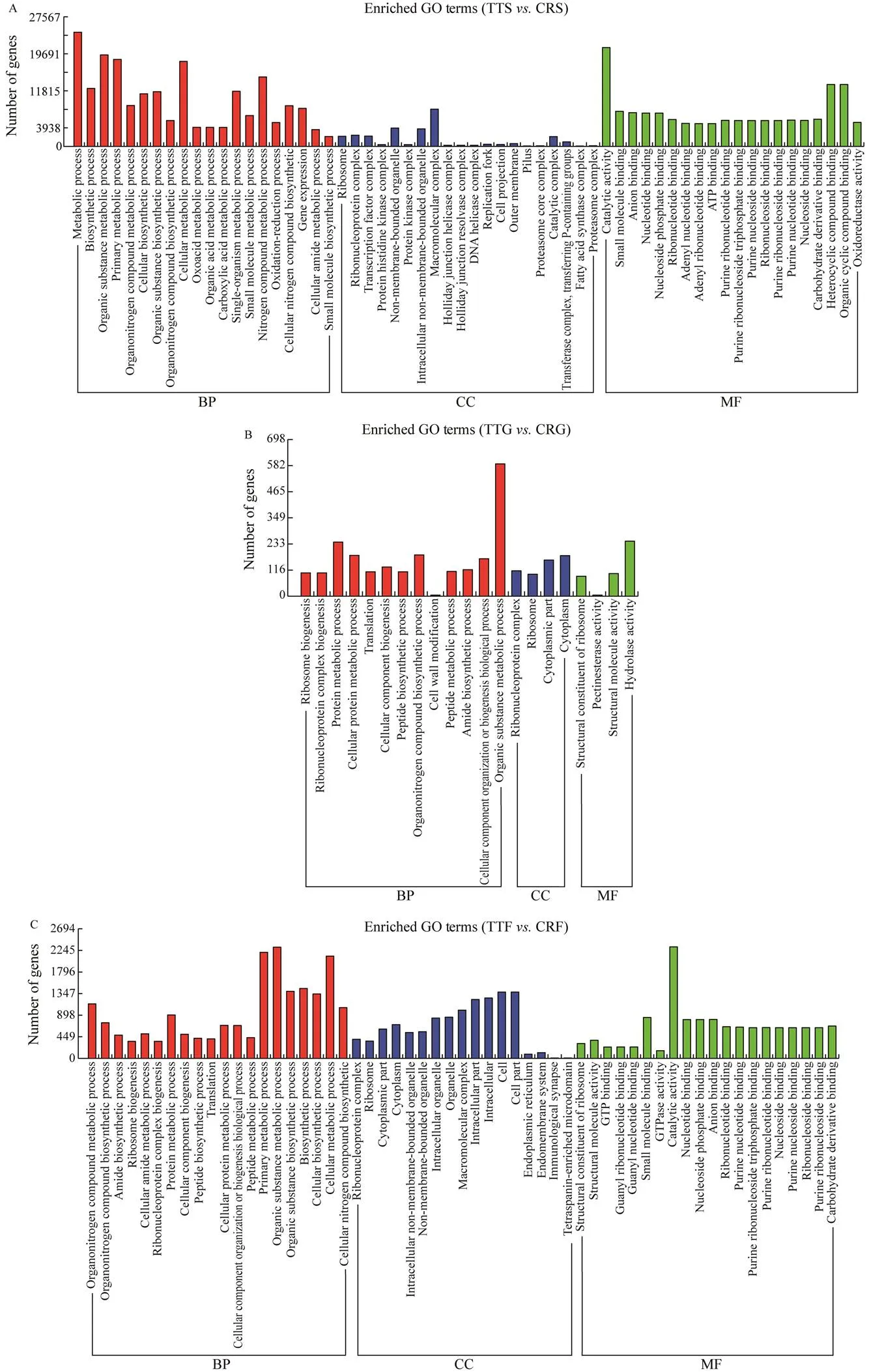
Fig.7 GO annotation of DEG functional classification. A, GO analysis of DEGs of TTS-CRS; B, GO analysis of DEGs of TTG-CRG; C, GO analysis of DEGs of TTF-CRF.
Among these GO terms, most DEGs in the TTS-CRS group involved in the biological process category were me-tabolic process (24283 genes), organic substance metabo-lic process (19471 genes), cellular metabolic process (18088 genes), primary metabolic process (18505 genes), and ni- trogen compound metabolism (14787 genes); most of the DEGs were classified into the cellular component and ma- cromolecular complex (7911 genes), while in the molecu- lar function category the corresponding annotation was in catalytic activity (21013 genes). For the DEGs in the TTG–CRG group, most were assigned to the biological process- es category: organic substance metabolic processes (589 genes), protein metabolism (241 genes), and organonitro- gen compound biosynthesis (184 genes). In the molecular function category, most of the DEGs were involved in hy- drolase activity (245 genes), and most that were assigned to the cellular component were in cell cytoplasm (181 genes).For the DEGs in the TTF–CRF group, most DEGs that wereassigned to the biological process category were in orga-nic substance metabolic process (2311 genes), primary me- tabolic process (2205 genes), and cellular metabolic pro- cess (2126 genes). In the molecular function category, mostof the DEGs were involved in catalytic activity (2319 genes); the following groups were assigned to the cellular compo- nent (1377 genes, GO:0005623) and cell part (1377 genes, GO:0044464). The DEGs of the stomach involved in the GO terms were much more frequent than those of the mid- gut and fecal mucosa, and DEGs had different distribution characteristics in the stomach than in the other two tissues. Most DEGs of the midgut were involved in hydrolase ac- tivity, protein metabolism and cell cytoplasm, while most of the DEGs of the stomach and fecal mucosa were in- volved in catalytic activity and primary metabolic process- es.
4 Discussion
Horseshoe crabs are ancient marine arthropods with a long evolutionary history. It is very possible that they havebenefited from their special anatomical structure and func- tion, especially the omnivorous feeding habits and effec-tiveinnate immunity. However, their genetic mechanisms underlying their ability to adapt to environmental changes during their long evolutionary history are still unclear. There is no doubt that as a typical consumer of the coastal and ma- rine ecosystems, the key factors for horseshoe crabs’ sur- vival include feeding and the related digestion and absorp- tion.
Here, we report the first transcriptome assembly for the stomach, midgut, and fecal mucosa ofandjuveniles using the Illumina RNA-seq plat- form. We obtained from 40.37 million to 52.95 million clean reads and from 6.06Gb to 7.94Gb of clean bases from the samples. The genome assembly resulted in 373069 uni- genes with an N50 of 1314, while the transcriptome assem- bly resulted in 736378 unigenes and an N50 of 2121. These 373069 unigenes could be assigned to the protein databases for annotation. Most unigenes were involved in important biological processes related to digestion, absorption, and transportation.
Some annotated genes involved in immune systems andnervous systems were also found in the digestive tract. For example, multiple genes were involved in the immunity-related JAK-STATsignaling pathway. The functions of theJAK-STATsignaling pathway include defense response toviruses, positive regulation of the immune response, signal transduction, response to drugs, oxidation-reduction reac- tions, and other important processes. Thus, the digestive tract of the horseshoe crab is a complex system with mul- tiple functions that play important roles in survival and de- velopment.
There were a large number of genes in the digestive tract of the horseshoe crab that were similar to those of fishes, amphibians, and reptiles, as identified by annotation in the NR, NT, KO, SwissProt, PFAM, GO, and KOG databases. This may be due to horseshoe crabs having similar living habits, anatomical structure, or function of digestive organs. The structure of the digestive tract of the horseshoe crab is related to its omnivorous habit, and the function of se-creting mucus is similar to that in other animals. The struc- ture and function of the digestive tract of the horseshoe crab are closely related to its omnivorous feeding habit.
The previous research on comparative genomics shows thatand the Atlantic horseshoe crabhave the most orthologues shared among horse-shoe crab species (Liao, 2019). In this comparison betweenandjuveniles, the differences in gene expression in the same tissues between the two species also provide potential molecular mecha- nisms with which to explore the niche differentiation of two co-occurring horseshoe crab species in southwestern China, and the mechanism of differential expression war- rants further investigation.
The DEG assay showed that most DEGs in the stomach transcriptome data of.were down-regulated, while the DEGs in the fecal mucosa trans- criptome were mostly up-regulated. The different transcrip- tome patterns between organs might be related to the di- gestion process during feeding. The digestive tract is di- vided into foregut, midgut, and hindgut. The stomach be- ing enlarged to store and grind food is the main charac- teristic of the foregut. The anatomical structure and func- tion are similar between the midgut and hindgut (Chen., 2016). They are related to the digestion of organic matter in food, absorption of the hydrophilic and hydro- phobic nutrients, the secretion of intestinal fluids, and the transportation of food through the digestive tract, which can help horseshoe crabs adapt to an omnivorous diet. The cell processes and activities in the midgut, stomach, and fecal mucosa can be different during digestion. Further investi- gation showed that most DEGs of the midgut were involved in hydrolase activity, protein metabolism, and cell cytoplasm, while most DEGs of the stomach and fecal mucosa were involved in catalytic activity and primary metabolic pro- cesses, and most DEGs of the stomach were assigned to the cellular component of macromolecular complexes. Most DEGs of the fecal mucosa were assigned to the cellular component and cell parts categories.
The transcriptome data of fecal mucosa of horseshoe crabs may serve as a reference for environmental DNA (eDNA) research on horseshoe crab conservation. The ana- lysis of eDNA has significant potential for monitoring horse- shoe crab populations, thus influencing conservation efforts. The eDNA technology is a good biological utility for con- servation monitoring without requiring the collection of the living horseshoe crabs. When individuals interact with the environment, DNA is expelled with the excretions and ac- cumulates in the surrounding environment. Sources of eDNA include the horseshoe crab’s feces, mucus, and shells. High-throughput DNA sequencing methods including meta- genomics, metabarcoding, and single-species detection can be used to analyze these samples for rapid monitoring and application to horseshoe crab conservation. The transcrip- tome data of fecal mucosa herein shows that it can be a good source of experimental materials for eDNA technol- ogy indicating environmental traces of horseshoe crabs.
The present study provides a better understanding of the adaptation of two horseshoe crab species to an omnivorous diet and presents an available transcriptome resource for future research. The horseshoe crabs require substantial foodresources, a suitable hydroclimate, and deeper water regionsof the bay and the adjacent continental shelf (Tanacredi, 2009). While the horseshoe crab’s life history with the distinct stages in complex habitats make it vulnerable to various threats at each stage of its development, horse- shoe crabs have survived on Earth for more than 450 mil- lion years. Therefore, most genes annotated from the di- gestive tract were assigned to key life processes including metabolism, biogenesis, signal transduction, transportation, immune response, regulatory, binding, catalysis, secretion, and environmental information signal processing. The di- gestive tract may also play a key role in host adaptation to complex and diverse environments and resistance to the in- vasion of pathogenic microorganisms in the multiple in- shore and estuarine environments. Highly effective adapta- tion significantly decreases the need for differentiated phe- notypic variants associated with environmental impacts, thereby providing the conditions for long-term evolution- ary success. The horseshoe crab is an ancient creature that is both simple and sophisticated regarding its ability to adapt to the changes in the global environment.
5 Conclusions
The transcriptome assemblies for the stomach, midgut, and fecal mucosa ofandju- veniles from the northern Beibu Gulf in March 2019 were reported here for the first time. The genome assembly re- sulted in 373069 unigenes with an N50 of 1314, while the transcriptome assembly resulted in 736378 unigenes and an N50 of 2121. The unigenes annotated from the diges- tive tract of horseshoe crabs showed the highest similarity to those of fishes, amphibians, and reptiles, and most uni- genes were closely related to the functions of metabolism, binding, translation, biogenesis, catalysis, signal transduc- tion, energy production, transportation, immune response, secretion, and environmental information signal process-ing.Compared with, nearly all of the (99.95%) DEGs in thestomach were down- regulated, while almost all (99.97%) of the DEGs offecal mucosa and about half (55.42%) of theDEGs of themidgut were up-regulated. Most DEGs of the midgut were involved in hydrolase activity, protein metabolism, and cytoplasm, while most DEGs of the stomach and fecal mucosa were involved in catalytic activity and primary metabolic processes, and most DEGs of the stomach were assigned to the cellular component of macromolecular complexes. Most DEGs of the fecal mu- cosa were assigned to the cellular component of cells and cell parts. The transcriptome data of the fecal mucosa can provide a reference for environmental DNA research for endangered species. The results will also facilitate the in- vestigation of molecular mechanisms of the digestive tract related to feeding habits and environmental traces of horse- shoe crabs.
Acknowledgements
This work was supported by the Scientific Research Pro-ject of Huaqiao University (No. 605-50X18005), the Guang- xi BaGui Youth Scholars Program, and the Guangxi Re- cruitment Program of 100 Global Expert.
Carmichael, R. H., Gaines, E., Sheller, Z., Tong, A., Clapp, A., and Valiela, I., 2009. Diet composition of juvenile horseshoe crabs: Implications for growth and survival of natural and cul-tured stocks. In:Tanacredi, J. T.,., eds., Springer, New York, 521-534, DOI: 10.1007/978-0-387-89959-6_33.
Chatterji, A., Vijayakumar, R., and Parulekar, A. H., 1988. Growthand morphometric characteristic in the horseshoe crab,(Latreille) from Canning (West Ben- gal), India., 31: 352-353.
Chen, C. P., Yang, M. C., Fan, L. F., Qiu, G., Liao, Y. Y., and Hsieh, H. L., 2015. Co-occurrence of juvenile horseshoe crabsandin an estuarine bay, southwestern China., 24 (2): 117-126, DOI: 10.3354/ab00641.
Chen, X. L., Zhu, W. L., Yang, C. L., Luo, B., Li, Q. Z., Peng, J. X.,., 2016. Morphological and histological study on the digestive tract ofand, 37 (5): 92-100, DOI: 10. 15928/j.1674-3075.2016.05.014 (in Chinese with English ab- stract).
Fan, L. F., Chen, C. P., Yang, M. C., Qiu, G., Liao, Y. Y., and Hsieh, H. L., 2017. Ontogenetic changes in dietary carbon sources and trophic position of two co-occurring horseshoe crab species in southwestern China.,26: 15-26, DOI: 10.3354/ab00670.
Gaines, E. F., Carmichael, R. H., Grady, S. P., and Valiela, I., 2002. Stable isotopic evidence for changing nutritional sources of juvenile horseshoe crabs., 203(2): 228-230, DOI: 10.2307/1543412.
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thomp- son, D. A., Amit, I.,., 2011. Full-length transcriptome as- sembly from RNA-Seq data without a reference genome.,29: 644-652, DOI: 10.1038/nbt.1883.
Hong, S. G., 2011.,. Xiamen University Press, Xiamen, 342pp(in Chinese).
Laurie, K., Chen, C. P., Cheung, S. G., Do, V., Hsieh, H., John, A.,., 2019.(errata version published in 2019).: e.T21309A149768986.
Liao, Y. Y., Xu, P. W., Kwan, K. Y., Ma, Z. Y., Fang, H. Y., Xu, J. Y.,., 2019. Draft genomic and transcriptome resources for marine chelicerate., 6: 190029, DOI: 10.1038/sdata.2019.29.
Novitsky, T. J., 2015. Biomedical implications for managing theharvest along the northeast coast of the United States, In:.Carmichael, R. H.,., eds., Springer, New York, 483-500, DOI: 10.1007/ 978-3-319-19542-1.
Smith, D. R., Brockmann, H. J., Beekey, M. A., King, T. L., Millard, M. J., and Zaldívar-Rae, J., 2016. Conservation sta- tus of the American horseshoe crab, (): A regional assessment., 27 (1): 135-175, DOI: 10.1007/s11160-016-9461-y.
Tanacredi, J. T., Botton, M. L., and Smith, D. R., 2009.. Springer, New York, 692pp, DOI: 10.1007/978-0-387-89959-6.
Van Roy, P., Orr, P. J., Botting, J. P., Muir, L. A., Vinther, J., Le- febvre, B.,., 2010. Ordovician faunas of Burgess Shale type.,465 (7295): 215-218, DOI: 10.1038/nature09038.
Wang, C. C., Kwan, K. Y., Shin, P. K. S., Cheung, S. G., Itaya, S., Iwasaki, Y.,., 2020. Future of Asian horseshoe crabconservation under explicit baseline gaps: A global perspec- tive.,24: e01373, DOI: 10. 1016/j.gecco.2020.e01373.
Wang, L., Yang, X., Zhou, S., Lyu, T., Shi, L., Dong, Y.,., 2021. Comparative transcriptome analysis revealed omnivorousadaptation of the small intestine of Melinae.,11 (1): 19162, DOI: 10.1038/s41598-021-98561-0.
Xie, M. J., Zhong, J. X., Xie, X. Y., Li, J. W., and Zhu, C. B., 2018. Histology and mucous cell distribution in digestive tract of.,53(2): 270-277, DOI: 10.13859/j.cjz.201802013.
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A., 2010. Gene ontology analysis for RNA-seq: Accounting for selection bias., 11 (2): R14, DOI: 10.1186/gb- 2010-11-2-r14.
Zhang, L., Xu, B., Wu, T., Yang, Y., Fan, L., Wen, M.,., 2017. Transcriptomic profiling of two Pak Choi varieties with contrasting anthocyanin contents provides an insight into struc- tural and regulatory genes in anthocyanin biosynthetic path- way.,18 (1): 288, DOI: 10.1186/s12864-017- 3677-7.
J. Ocean Univ. China(Oceanic and Coastal Sea Research)
https://doi.org/10.1007/s11802-022-5321-7
ISSN 1672-5182, 2022 21 (3): 591-600
(January 4, 2022;
March 14, 2022;
April 5, 2022)
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2022
Corresponding author. E-mail: liyuh@hqu.edu.cn
(Edited by Qiu Yantao)
 Journal of Ocean University of China2022年3期
Journal of Ocean University of China2022年3期
- Journal of Ocean University of China的其它文章
- Effect of Intertidal Elevation at Tsuyazaki Cove, Fukuoka,Japan on Survival Rate of Horseshoe Crab Tachypleus tridentatusEggs
- Asian Horseshoe Crab Bycatch in Intertidal Zones of the Northern Beibu Gulf: Suggestions for Conservation Management
- Experimental Investigation on the Interactions Between Dam-Break Flow and a Floating Box
- Variational Solution of Coral Reef Stability Due to Horizontal Wave Loading
- High Microplastic Contamination in Juvenile Tri-Spine Horseshoe Crabs: A Baseline Study of Nursery Habitats in Northern Beibu Gulf, China
- Influence of Autonomous Sailboat Dual-Wing Sail Interaction on Lift Coefficients