
ATF6对骨肉瘤MCA205细胞免疫原性的调控作用及其机制的初步探索
2022-10-29 08:27黄恩厚李莫寒李培培夏琳马瑜婷南京医科大学肿瘤个体化医学协同创新中心江苏南京211166中国医学科学院系统医学研究院北京100005苏州系统医学研究所江苏苏州215123
中国肿瘤生物治疗杂志 2022年8期
黄恩厚,李莫寒,李培培,夏琳,马瑜婷(1.南京医科大学 肿瘤个体化医学协同创新中心,江苏 南京 211166;2.中国医学科学院 系统医学研究院,北京 100005;3.苏州系统医学研究所,江苏 苏州 215123)
乳腺癌、胰腺癌、肺癌、皮肤癌、前列腺癌等多种实体肿瘤细胞中均可观察到内质网应激(ER stress,ERS)[1],在晚期或化疗抵抗的肿瘤中尤为明显[2]。未折叠蛋白反应(unfolded protein response,UPR)是高度保守的ERS 响应机制。在UPR 的3 条分支通路中,活化转录因子6(activating transcription factor 6,ATF6)的研究进展相对缓慢。正常状态下,ATF6 与葡萄糖调节蛋白78(glucose-regulated protein 78,GRP78)结合。当发生ERS 时,大量错误折叠的蛋白可挤占结合GRP78,释放出游离的ATF6[3]。ATF6 从ER转移到高尔基体后,可被蛋白酶切割,产生含有碱性亮氨酸拉链(basic leucine zipper,bZIP)的转录因子,增强分子伴侣蛋白、折叠酶及内质网相关蛋白质降解效应分子的表达[4-5],有利于维持蛋白质稳态和细胞存活。
ATF6 通路的持续活化可帮助癌细胞抵抗ERS 引发的细胞死亡[6-8]、支持休眠癌细胞存活[9]、促进化疗抵抗[10]、诱导上皮细胞间质化[11],以及增强血管新生[12]。也有报道[13]显示,ATF6 是决定C/EBP同源蛋白(C/EBP homologous protein,CHOP)表达的早期转录因子,因此也具有促凋亡活性。浸润肿瘤的髓系细胞也会发生ERS 和UPR,可造成免疫抑制和免疫耐受,促进炎症,阻碍抗肿瘤免疫应答[14-15]。条件性敲除髓系细胞的Atf6后,肿瘤内多核型髓源抑制性细胞的免疫抑制功能显著下降,肿瘤进展明显延缓[16]。目前,ATF6 通路对癌细胞免疫原性(即肿瘤细胞被免疫系统识别和清除的能力)的影响尚属未知[17]。本研究拟借助免疫健全和免疫缺陷小鼠模型,探索野生型(WT)和Atf6-/-癌细胞的免疫原性和体内成瘤能力的差异,并初步挖掘相关调控机制。
1 材料与方法
1.1 主要材料与试剂
C57BL/6N 和nu/nu小鼠(购自北京维通利华实验动物技术有限公司,实验动物合格证号:20220107Abzz0619000267、20220107Abzz061900076 5、20201209 Abzz0619000481)与 B6.129S2-Ifnar1tm1Agt/Mmjax 小鼠(Ifnar-/-,Jackson 实验室)的饲养和实验均在中国医学科学院系统医学研究院/苏州系统医学研究所的SPF级实验动物中心完成,实验方案已获得苏州系统医学研究所实验动物使用与管理委员会的批准(批准号:ISM-IACUC-0072-R)。小鼠按随机数字表法随机分组,每组小鼠至少5 只,年龄和性别匹配,均为雌性,6~7 周龄,体质量为18~20 g。骨肉瘤细胞MCA205由法国国家健康与医学研究院Guido Kroemer实验室馈赠。表达ISRE-荧光素酶的成纤维细胞L929-ISRE 由北京大学蒋争凡实验室馈赠。293FT 细胞购自Thermo Fisher 公司。所有细胞均培养于含有L-谷氨酰胺和HEPES的高糖DMEM 培养基[BL304A 购自Biosharp 公司,额外添加含10% 胎牛血清(FBS-12A,购自Capricorn Scientific 公司)和100 U/mL 的青霉素和链霉素双抗溶液(BL505A,购自Biosharp 公司]。LentiCRISPRv2(52961)、psPAX2(12260)和pMD2.G(12259)质粒均购自Addgene公司。
细胞转染采用的FuGENE®HD Transfection Reagent(E2311)购自Promega 公司,聚凝胺(polybrene,107689)购自MERCK 公司,嘌呤霉素(ant-pr)购自InvivoGen 公司,细胞活力检测试剂盒CKK-8(BS350A)购自Biosharp 公司,能量代谢检测试剂盒Seahorse XF Glycolysis Stress Test Kit(103020-100)和Cell Mito Stress Test Kit(103015-100)均购自Agilent Technologies公司,细胞死亡检测使用的PE Annexin V(640947)和DAPI(564907)分别购自BioLegend 公司、BD BioScience 公司,衣霉素(tunicamycin,Tm;654380)购自Sigma-Aldrich 公司。离子霉素(ionomycin;ab120116)和Rhod-4 染料(ab112157)均购自Abcam公司,组织消化酶Liberase™TL(05401020001)购自Roche 公司,DNase Ⅰ(260913)购自Millipore 公 司,IFN-γ 的ELISPOT 试剂盒(551083)和ELISA 试剂盒(430801)分别购自 BD Bioscience 和 BioLegend 公司,Dual-Luciferase®Reporter 试剂盒(E1960)、胞内ATP 检测试剂盒(G7571)均购自Promega 公司,胞外ATP 检测试剂盒(FLAA)购自Sigma-Aldrich公司,组织和细胞总RNA提取试剂盒(DP419)购自TIANGEN公司,逆转录试剂盒(RR047A)和基于SYBR Green 的qPCR试剂盒(RR820A)均购自TaKaRa 公司,CD90.2 抗体(105310)购自BioLegend 公司,Low-Tox®-M 兔补体(Cl3051)购自Cedarlane公司,IL-2(5 000 U/mL)购自江苏四环生物制药公司,荧光染料eFlour670(65-0840-85)购自eBioscience公司。
1.2 CRISPR-Cas9敲除MCA205细胞的Atf6基因
靶向Atf6基因的gRNA 序列为5'-TCG ACGTTGTTTGCTGAACT-3',合成正义和反义寡核苷酸,退火形成双链DNA 片段,连接至线性化的LentiCRISPRv2 载体。上述gRNA 载体质粒、包装质粒psPAX2和pMD2.G按照21∶15∶6的比例共同转染293FT 细胞。48 h后,收集病毒上清液并添加4 μg/mL聚凝胺后感染MCA205细胞。感染后48 h,用4 μg/mL嘌呤霉素筛选5 d。从存活的细胞中分选单克隆并逐一测序鉴定。
1.3 CCK-8法检测Atf6缺失对MCA205细胞活力的影响
将WT 或Atf6-/-MCA205 细胞接种在96 孔板中(每孔2×103个细胞、100 μL 培养基),用PBS 或12 μmol/L 的Tm 处理细胞,在指定时间点向培养基中加入CCK-8 试剂(10 μL),借助SpectraMax®i3x 多功能微孔读板仪检测450 nm 和600 nm 处的光密度(D)值,绘制细胞增殖或死亡曲线。
1.4 细胞能量代谢仪检测Atf6缺失对MCA205细胞OCR和ECAR的影响
在XF-24 板中接种细胞(5×104个/孔)并培养过夜,用12 μmol/L 的Tm 预处理细胞8 h,检测前将培养基更换为Seahorse XF 基础培养基,37 ℃培养1 h。借助Seahorse XFe24 分析仪,按照试剂盒说明,依次注射10 mmol/L 葡萄糖(GLU)、1 μmol/L 寡霉素(Oligo)和100 mmol/L 的2-脱氧-D-葡萄糖(2-deoxy-D-glucose,2-DG)后检测细胞ECAR。依次注射1.5 μmol/L 寡霉素,1 μmol/L 线粒体氧化磷酸化解偶联 剂(carbonylcyanide-p-trifluoromethoxyphenyl hydrazine,FCCP)和0.5 μmol/L 鱼藤酮/抗霉素(Rot/AA)后检测细胞OCR。检测完成后立即消化获取每孔细胞,严格计数后用于数据的归一化处理。
1.5 流式细胞术检测Atf6缺失对MCA205细胞死亡的影响
在24 孔板中接种WT 或Atf6-/-MCA205 细 胞(1×105个/孔)。待细胞汇合度达到70%~80%时,用PBS 或12 μmol/L 的Tm 处理细胞16 h,Annexin V 和DAPI 染色后用流式细胞仪(BD LSRFortessa™)分析ERS诱导的细胞死亡。
NK细胞体外培养与杀伤检测:收集新鲜骨髓细胞,借助红细胞裂解液、CD90.2 抗体和Low-Tox®-M兔补体去除红细胞和T 细胞。余下的细胞在含有5 000 U/mL IL-2 和10% FBSR 的PMI1640 培养基中培养7 d,每天半量更换培养基。如此获得的NK 细胞分别与eFlour670预染的WT或Atf6-/-MCA205细胞按照不同的效靶比混合,共培养4 h 后借助Annexin V和DAPI染色检测肿瘤细胞死亡。流式检测结果用FlowJo软件分析。
1.6 构建MCA205细胞小鼠移植瘤模型
在C57BL/6N、nu/nu或Ifnar-/-小鼠背部两侧皮下标注部位同时分别注射1×106个WT或Atf6-/-MCA205细胞,每隔2~3 d测量并记录肿瘤的最大直径及其垂直方向的肿瘤直径,用两者的乘积代表肿瘤大小,绘制生长曲线。
1.7 移植瘤组织及肿瘤细胞的基因转录水平分析
植瘤后7 d 剥离和收集肿瘤组织,获取PBS或Tm(12 μmol/L)处理16 h的肿瘤细胞,分别提取样本总RNA,NanoDrop™2000 分光光度计确定其浓度和纯度。肿瘤转录组测序委托苏州金唯智生物科技公司,由Illumina HiSeq X Ten 平台完成。借助R 语言程序包(DESeq2、FactoMineR、ComplexHeatmap 和EnhancedVolcano)分析基因表达,绘制主成分分析、热图和火山图。Atf6-/-较WT 肿瘤组织中显著上调基因的GO富集分析由Metascape在线工具完成。肿瘤细胞mRNA 被逆转录为cDNA 后,可借助如下引物(表1)对特定基因转录水平进行PCR检测。
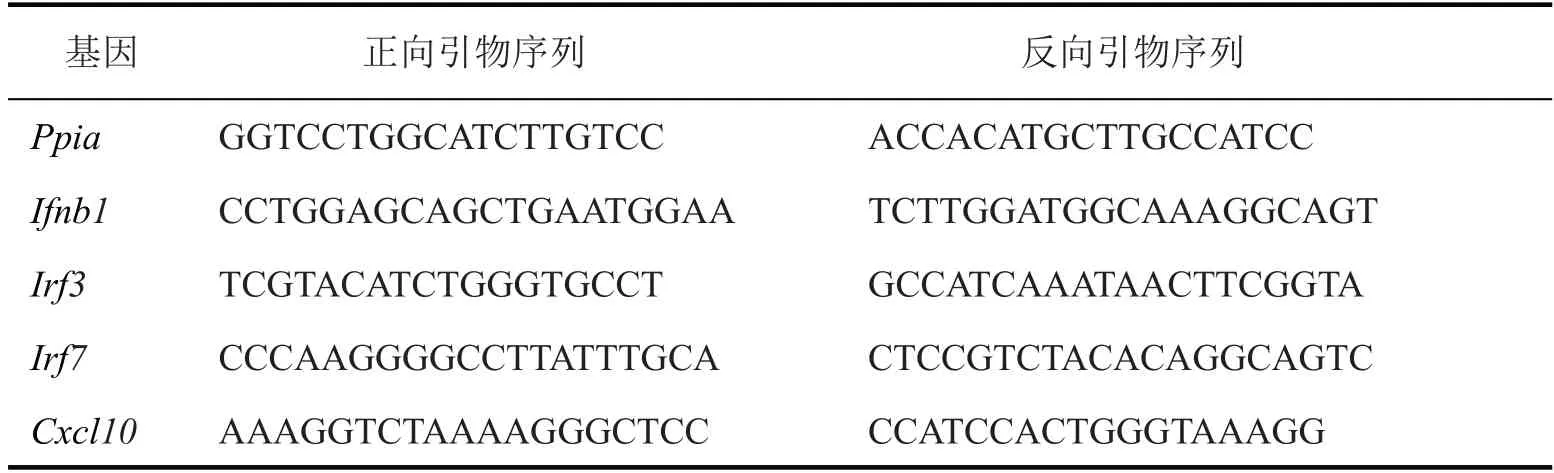
表1 qPCR所用引物序列
1.8 ELISPOT 及ELISA 实验检测Atf6 缺失对效应T细胞活化的影响
小鼠皮下接种癌细胞7 d后,收集肿瘤组织并剪碎,用Liberase TL(0.4 Wünsch units/mL)和DNaseⅠ(200 U/mL)消化30 min,70 μm滤器滤过后制备单细胞悬液。在预先包被了IFN-γ 捕获抗体的多孔滤膜板中分配细胞悬液(1×106个/孔),培养16 h,反复清洗后,借助IFN-γ 检测抗体、辣根过氧化物酶(HRP)和显色底物,依托CTL-Immunospot S6酶联免疫斑点分析仪对肿瘤内分泌IFN-γ 的细胞进行定量分析。将Tm 预处理的WT 或Atf6-/-MCA205 细胞注射到小鼠后足掌皮下,7 d 后收集腘窝(引流)淋巴结制备单细胞悬液,上述细胞(1×106个)与Tm预处理的WT型MCA205 细胞(2×104个)共培养72 h,收集上清液并用ELISA试剂盒检测IFN-γ丰度。
1.9 流式细胞术检测Atf6缺失对胞内钙离子动员的影响
将WT和Atf6-/-MCA205细胞接种于黑色光学底透微孔板(2×104个/孔),PBS或Tm(12 μmol/L)处理16 h后,用不含Ca2+的HBSS缓冲液制备Rhod-4染料溶液并与待测细胞混合,室温下放置1 h。借助SpectraMax®i3x(激发波长540 nm,发射波长590 nm),先记录本底信号30 s,加入离子霉素(10 μmol/L)刺激后每5 s收集一次信号,绘制钙离子动员曲线。
1.10 化学发光法检测Atf6 缺失对胞内外ATP 浓度的影响
Tm 处理WT 或Atf6-/-MCA205 细胞16 h,收集细胞上清液,梯度稀释ATP 标准品,将50 μL 细胞上清液、标准品转移到全白96孔板,与等体积的检测工作液混合后立即用SpectraMax®L定量检测化学发光强度,计算胞外ATP浓度。为检测胞内ATP浓度,保留板孔底部细胞及100 μL 细胞上清液,加入等体积检测工作液,室温振荡10 min,吸取100 μL混合液转移至全白96孔板,用SpectraMax®L完成定量分析。
1.11 ISRE-荧光素酶报告细胞实验检测Atf6缺失对IFNα/β分泌的影响
将L929-ISRE 细胞接种于96 孔板中(5×104个/孔)过夜,加入Tm 预处理的WT 或Atf6-/-MCA205 细胞(1×105个/孔),共培养4 h 后弃上清液,加入80 μL裂解液充分震荡30 min,取其中50 μL,采用Dual-Luciferase®Reporter 试剂盒(Promega,E1960)由SpectraMax®L测定IFNα/β分泌水平。
1.12 WB检测Atf6缺失对肿瘤内死亡相关通路活化的影响
用含有蛋白酶抑制剂、磷酸酶抑制剂(A32959,购自Thermo Fisher Scientific 公司)的裂解液(P0013,购自Beyotime 公司)提取WT 和Atf6-/-MCA205 肿瘤组织的总蛋白,与上样缓冲液混合后煮沸变性。用12.5%SDS-PAGE分离蛋白样本,将蛋白印记电泳条带转移至硝酸纤维素(NC)膜,与如下抗体(1∶1 000稀释)4 ℃反应过夜:GSDMD(ab209845)和GSDME(ab21519,购于Abcam 公司)、Caspase3(14220)和cleaved caspase3 抗 体(9661,购 自Cell Signaling Technology 公 司)。β-actin(HRP-60008,购 自Proteintech公司)用作内参对照。之后,NC膜与HRP标记的二抗室温反应2 h,反复洗涤后用Amersham™ECL™Prime 试剂(RPN2232,GE 公司)在ChemiDoc™成像系统(Bio-Rad公司)中进行半定量分析。
1.13 ATF6基因表达与肿瘤临床预后的相关性分析
借助TCGA 数据库,分析ATF6表达水平与多种癌症患者总体生存率的关联,绘制Kaplan-Meier生存曲线,计算风险比(HR)、95%置信区间(CI)和Logrank检验P值。
1.14 统计学处理
采用GraphPad Prism 8 软件进行数据作图和统计学分析。实验至少有3个平行重复,所有实验至少独立重复2 次,计量数据以平均值±标准误(SEM)来表示。对于体内成瘤实验,首先计算每只小鼠肿瘤生长曲线下面积(AUC),之后分析不同组别小鼠AUC间的差异。采用Log-rank检验分析不同组患者Kaplan-Meier 生存曲线的差异。其他数据均采用双尾非参数Mann-WhitneyU检验。P<0.05则表示差异具有统计学意义。
2 结果
2.1 Atf6缺失对MCA205细胞生物学特性的影响
借助CRISPR-Cas9 技术对MCA205 骨肉瘤细胞的Atf6进行基因编辑,通过多轮DNA 测序筛选出基因序列的缺失的细胞单克隆(图1A)。CCK-8法检测结果(图1B)显示,WT 和Atf6-/-细胞在体外的活力和增殖速度相当;Tm预处理16 h,两种细胞的活力均受到明显抑制,且撤药后两者的活力均无法恢复。流式细胞术分析结果(图1C)显示,Tm 处理后,Atf6-/-细胞死亡比例低于WT 细胞(P<0.01)。细胞能量代谢分析结果(图1D、E)显示,Tm 预处理后,WT 和Atf6-/-细胞的糖酵解(用ECAR 表征)和氧化磷酸化(用OCR表征)水平没有明显差异。内质网Ca2+动员是高度灵敏的ERS 标志物,且与细胞死亡密切相关[18-19],可借助Rhod-4 对其进行实时监测。本研究发现,对经PBS及Tm预处理的肿瘤细胞,离子霉素可快速诱导其胞内Ca2+动员,之后逐步恢复稳态,而上述过程不受Atf6缺失的影响(图1F)。上述结果提示,Atf6缺失不影响PBS和Tm处理后肿瘤细胞的活力和增殖、能量代谢、以及离子霉素引发的内质网Ca2+动员,但可降低Tm诱导的细胞死亡。

图1 Atf6对肿瘤细胞活力、增殖、死亡、能量代谢和钙动员的影响
2.2 Atf6缺失可增强MCA205骨肉瘤的免疫原性
在免疫健全的C57BL/6N 小鼠皮下接种WT 或Atf6-/-骨肉瘤细胞,可观察到WT 肿瘤迅速生长,而Atf6-/-肿瘤的生长则明显滞后(图2A)。出乎意料的是,在免疫缺陷的nu/nu小鼠(T 细胞发育受阻)背部两侧皮下同时接种这两种癌细胞时,虽然Atf6-/-肿瘤的生长仍略慢于WT 肿瘤,但是两者均可快速生长(图2B)。上述现象提示,免疫健全小鼠体内Atf6-/-肿瘤的滞长与T细胞激活密切相关。C57BL/6N小鼠接种癌细胞后第7天,WT与Atf6-/-肿瘤大小刚刚显示出差异,收集此时的肿瘤组织并进行转录组测序,随后进行主成分分析并绘制差异基因热图和火山图,结果都显示,两者的基因转录谱图存在明显差异(图2C、E)。GO 富集分析结果(图2F)提示,与WT 肿瘤相比,Atf6-/-肿瘤微环境中免疫防御应答、细胞因子分泌、炎症反应、免疫细胞活化和迁移、Ⅰ型和Ⅱ型IFN应答、细胞杀伤相关的基因表达均显著增强。ELISPOT 实验结果(图2G)显示,与WT 肿瘤相比,Atf6-/-肿瘤内分泌IFN-γ 的抗肿瘤效应细胞显著增多(P<0.01)。将经Tm 预处理的WT 与Atf6-/-癌细胞注射到C57BL/6N小鼠后足掌皮下,可刺激肿瘤抗原特异性T 细胞,使其在引流淋巴结(draining lymph node,DLN)中初次活化(prime)。收集上述DLN 并制备混合细胞悬液,用Tm 预处理的WT 癌细胞在体外再次刺激(boost),可诱导肿瘤抗原特异性T 细胞的再次活化并分泌IFN-γ。检测结果(图2H)显示,与对照组相比,被Tm预处理的Atf6-/-癌细胞初次活化过的小鼠,其DLN 内T 细胞经体外再次刺激后,IFN-γ分泌水平显著升高(P<0.001)。在体外按照1∶1 和5∶1 的效靶比将NK 细胞分别与WT 与Atf6-/-癌细胞共培养,结果(图2I)显示,Atf6-/-癌细胞比WT 癌细胞更易被NK 细胞杀伤(均P<0.05)。上述结果提示,Atf6缺失可增强肿瘤的免疫原性,促进效应T 细胞和NK细胞的免疫监视功能,塑造出抗肿瘤的免疫微环境。
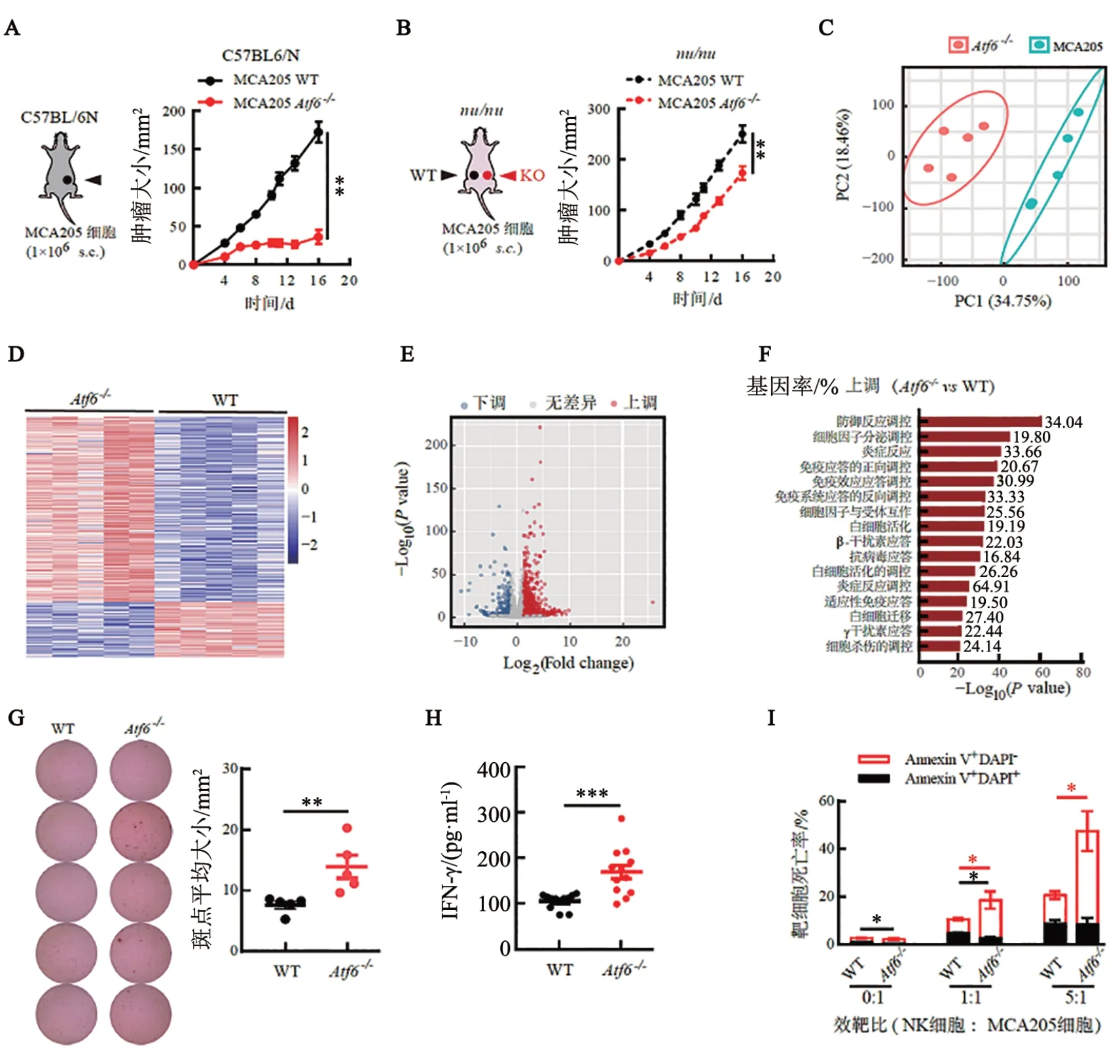
图2 Atf6缺失可增强MCA205细胞移植瘤的免疫原性
2.3 影响Atf6-/-MCA205细胞免疫原性的可能因素
细胞应激后释放的“危险信号分子”ATP 和IFNα/β 是决定肿瘤免疫原性的重要因素[20-21]。Tm 刺激16 h 可诱导细胞发生ERS。本研究分别检测了WT与Atf6-/-MCA205 细胞的胞内ATP、释放至胞外的ATP及IFN-α/β 的分泌水平,均未发现明显差异(图3A~C)。因此,Atf6缺陷并不影响MCA205 细胞在ERS 状态下释放ATP 和IFN-α/β。基因表达分析结果(图3D)显示,Tm 刺激下Irf7和Irf3转录本的丰度在Atf6-/-细胞内显著增加(均P<0.001)。Irf7和Irf3是决定IFN-α/β 及诸多ISG 表达的关键转录因子基因。Irf3在IFN-α/β 转录的启动阶段发挥了核心作用,但其蛋白可被快速降解。IFN-α/β 与其受体IFNAR 结合后,可促进Irf7的转录,进一步增强IFN-α/β 的表达。因此,Irf7密切参与了IFN-α/β的正反馈调控[22]。本研究提示,ATF6 可抑制ERS 触发的Irf7和Irf3表达。Tm 刺激后,虽然两种细胞内Ifnb和Cxcl10转录本的丰度相当(图3D),但是Irf7和Irf3在Atf6-/-癌细胞内的上调仍然可能促进IFN-α和多种ISG的表达,而这可能会增强肿瘤细胞的免疫原性。此外,本研究还探索了宿主因素对Atf6-/-肿瘤免疫原性的影响,重点关注抗肿瘤的效应T 细胞和IFNAR 信号通路[23-25]。虽然ATF6 缺陷型肿瘤在免疫健全小鼠体内会出现滞长,甚至自发消退,但其在缺乏T细胞的nu/nu小鼠体内可快速生长。接种于Ifnar-/-小鼠皮下的Atf6-/-肿瘤仍然会出现滞长和自发消退,但与接种在WT小鼠皮下的Atf6-/-肿瘤相比,其开始消退的“拐点”时间会明显推迟,肿瘤生长的峰值也会更大(图3E)。上述结果提示:机体免疫系统监视和清除Atf6-/-肿瘤的过程中,T 细胞是关键的效应细胞,而宿主的IFNAR信号通路也发挥了一定作用。本研究还借助WB法,比较了WT和Atf6-/-肿瘤组织内细胞死亡相关分子的表达水平和活化程度。Atf6-/-肿瘤组织中的caspase3 总量、切割后活化的caspase3 的丰度都明显低于WT 肿瘤。作为介导焦亡的关键分子,焦孔素GSDME 和GSDMD 在WT 和Atf6-/-肿瘤组织中的表达水平相当,且两者N 端片段的切割均未能检测到(图3F)。上述结果提示,Atf6缺失肿瘤的滞长和消退并非由于凋亡或焦亡通路的增强,更可能是因为抗肿瘤免疫应答的激活。
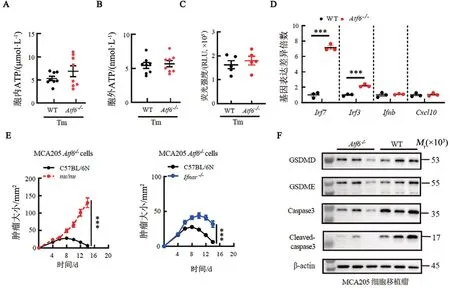
图3 Atf6-/-MCA205细胞免疫原性的可能影响因素
2.4 ATF6低表达的肿瘤患者预后更好
为了验证小鼠肿瘤模型中上述发现是否具有临床相关性,本研究对肿瘤公共数据库进行了挖掘。在肺鳞癌(495例患者)、头颈鳞癌(363例患者)、宫颈鳞癌(304例患者)、膀胱癌(404例患者)队列中,均发现ATF6表达低患者的总体生存期更长(图4)。
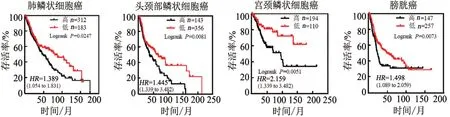
图4 肿瘤ATF6表达水平的临床预后价值分析
3 讨论
肿瘤发生、发展和治疗过程中,多种不利的微环境因素可造成癌细胞的稳态失衡、激活自噬、UPR、氧化应激等高度保守的细胞应激反应,通过降解自身组分产生营养物质、减缓转录翻译、降解错误折叠蛋白和抑制脂质过氧化物生成等,缓解癌细胞的生存压力,帮助其存活、扩增及产生治疗抵抗。当不利刺激因素持续存在,或应激反应不足以减弱或逆转细胞损伤时,凋亡、程序性坏死、焦亡和铁死亡等信号通路可被激活,启动细胞的死亡程序[26]。
近年来,随着“免疫原性细胞死亡”(immunogenic cell death,ICD)概念的提出,细胞应激和死亡通路对肿瘤免疫原性的影响成为新兴的研究热点。多项研究证实:放化疗导致肿瘤细胞自噬、UPR、凋亡、程序性坏死,上述信号通路的激活可触发危险信号分子的释放,对增强肿瘤细胞的免疫原性至关重要。其中,ATG5、ATG7、caspase、RIP3或MLKL的缺失或阻断可抑制ATP的释放,RIP3或MLKL的缺失可降低HMGB1的释放,均不利于抗肿瘤免疫的启动[27]。化疗还可促进STAT3的磷酸化,该信号通路的活化不仅可以促进癌细胞的存活,还能抑制其Ⅰ型干扰素应答,阻碍抗肿瘤免疫的活化[28]。
UPR对肿瘤细胞免疫原性的影响仍然存在较大的认知空白。化疗可激活癌细胞的蛋白激酶R 样内质网激 酶(protein kinase-like ER-resident kinase,PERK)通路,该UPR 分支可促进钙网蛋白(calreticulin,CALR)由ER 转移到细胞膜外表面,增强癌细胞的免疫原性,帮助DC 摄取和提呈肿瘤抗原,激活效应T 细胞[29]。本研究重点关注ATF6 信号通路对肿瘤细胞免疫原性的影响,及其对机体自发(并非治疗引发)的免疫监视功能的调控作用。虽然敲除Atf6不影响MCA205 细胞在体外的增殖和能量代谢,但是Atf6-/-MCA205 细胞在免疫健全小鼠体内的成瘤速度显著降低,可出现滞长甚至自发消退。Atf6-/-肿瘤MCA205 细胞在Ifnar-/-小鼠体内仍然会出现滞长和自发消退,但其生长峰值和彻底消退所需时间均高于其在免疫健全小鼠内的对应参数。在nu/nu小鼠体内,WT 和Atf6-/-MCA205 细胞均可快速成瘤,两者生长的差异显著降低。值得注意的是,与WT 肿瘤相比,Atf6-/-移植瘤组织内凋亡、焦亡通路的活化并未增强,甚至还有一定程度的减弱。上述现象提示,Atf6-/-MCA205 细胞体内成瘤减缓并非依赖于凋亡或焦亡通路的活化,其主要原因是效应T细胞的激活,肿瘤宿主的IFNAR 信号通路也发挥了一定的促进作用。肿瘤组织的转录谱图分析提示,ATF6的缺失引发了肿瘤免疫微环境的重塑,IFN-α/β 相关应答、IFN-γ 通路、NK 细胞杀伤力、免疫细胞的活化和迁移均显著增强。ELISPOT实验也证实Atf6-/-肿瘤微环境中分泌IFN-γ的效应T细胞的丰度明显提升。
借助Tm预处理模拟不利微环境因素的刺激,可观察到Ⅰ型IFN应答的关键转录因子Irf7和Irf3的表达在Atf6-/-MCA205细胞中显著高于WT细胞。诱导肿瘤细胞ERS 后用于prime-boost 实验,发现Atf6缺失可促进肿瘤抗原特异性T 细胞的活化,增强其IFN-γ 分泌。此外,Atf6-/-MCA205 细胞对NK 细胞的杀伤更敏感。上述现象表明,ATF6是限制肿瘤细胞免疫原性的重要瓶颈。具体的分子机制有待在后续研究中深入挖掘。ATF6 是分子伴侣、折叠酶、ERAD 相关基因表达的重要调控因子[5],其缺失可能加剧蛋白错误折叠,引发新生抗原表位的暴露。此外,IFN-α/β 和IFN-γ 可促进Ⅰ类和Ⅱ类MHC 分子的表达[30-31],增强抗原的交叉提呈[32],有助于增强肿瘤抗原的提呈。在黑色素瘤中,IRE1α-XBP1 和PERK 通路的活化可 能会抑制MICA、MICB、B7H6等应激诱导配体分子的表达,从而抑制NK细胞的杀伤功能。Atf6是否也能发挥类似的调控作用值得进一步探索。总之,敲除Atf6不仅能降低肿瘤细胞抵御不利环境的存活能力,还能增强其免疫原性,通过激活抗肿瘤免疫应答有效预防和治疗肿瘤。对于多个癌种而言,ATF6低表达的患者,其总体生存时间更长。因此,针对该靶点开发抑制剂分子具有较好的应用前景。
猜你喜欢
中国动物检疫(2022年10期)2022-10-14
新传奇(2022年32期)2022-08-19
当代陕西(2022年5期)2022-04-19
中国科学探险(2021年2期)2021-06-01
科学与财富(2021年33期)2021-05-10
保健与生活(2020年6期)2020-03-20
恋爱婚姻家庭·养生版(2019年1期)2019-01-25
中国卫生产业(2018年12期)2018-05-14
中学课程辅导·教师教育(上、下)(2016年17期)2016-12-17
文理导航(2016年30期)2016-11-12
