
引入遗传因子的骨密度机器学习回归模型研究
2022-11-01 01:44陈鹏丽孔祥勇林勇
中国骨质疏松杂志 2022年10期
陈鹏丽 孔祥勇 林勇
上海理工大学医疗器械与食品学院,上海 200093
骨质疏松症是一种常见的复杂疾病,受到多种因素的影响,包括临床危险因素(个人的年龄、性别、体重、身高、饮食、既往骨折史、长期使用糖皮质激素等)以及遗传因素[1]。有多项研究[2-4]证明,遗传因素对骨密度(bone mineral density,BMD)具有显著影响,骨密度差异的遗传率在50 %~85 %之间。全基因组关联研究(genome-wide association study, GWAS)及荟萃分析(Meta-analysis)发现,相较只使用临床危险因素数据,加入骨密度相关的遗传变异因素可以显著提高骨折预测的准确度[5]。
目前,全基因组关联研究和荟萃分析已经发现了许多与骨密度、骨质疏松症和骨质疏松性骨折相关的位点[6-7]。然而复杂疾病的多因素性质使得传统的基于统计学分析方法的GWAS效果有限[8]。近年来研究者们[8]提出多种用于检测骨质疏松症致病因素的机器学习模型和方法,在对多种复杂疾病建模以及易感位点识别中取得了突出的成果。
但相关研究大多数只以单一因素对骨质疏松症进行分析,很少考虑遗传因素和临床因素特征之间的相互作用,且缺乏识别在生物学上具有可解释性的易感位点的能力。
本文提出了一种基于机器学习的骨质疏松症致病因素分析方法,引入遗传因素及临床风险因素,识别影响个体对骨质疏松易感性的最优特征组合。研究采用两阶段的特征选择方法,首先通过最大互信息系数(maximal information coefficient, MIC)筛选出与预测变量高度相关的SNP子集,然后将所选子集混合临床风险因素作为输入进行序列特征选择(sequential feature selection, SFS)。下面将对数据预处理、两阶段特征选择和预测的过程进行详细描述,并以模型均方根误差(root mean square error, RMSE)为指标,在不同的回归模型中进行对比分析,衡量这一方法的性能。
1 材料和方法
1.1 实验数据
本研究中选取的实验样本为2 263例白种人,包括555名男性和1 708名女性。该研究得到相关机构审查委员会批准,所有研究参与者在进入项目前都签署了知情同意书[9]。
研究使用全身骨密度值作为因变量,将对骨密度的影响因素划分为临床风险因素和遗传因素。以数据较全的年龄、性别(男性:1,女性:2)、体重、身高、体质量指数(BMI)作为临床风险因素;遗传因素为单核苷酸多态性(SNP)基因分型数据。受试者临床风险因素基本信息见表1。
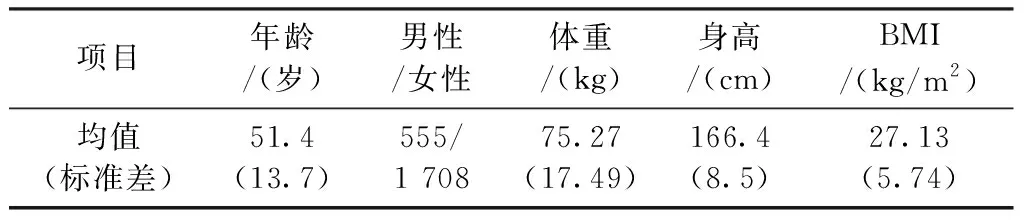
表1 受试者临床风险因素基本信息Table 1 Basic information of clinical risk factors of subjects
1.2 引入遗传因素的机器学习骨密度回归模型
本文提出的对骨密度回归模型的分析流程如图1所示,包括4个阶段:①数据预处理;②特征选择;③建立预测模型;④模型评估。
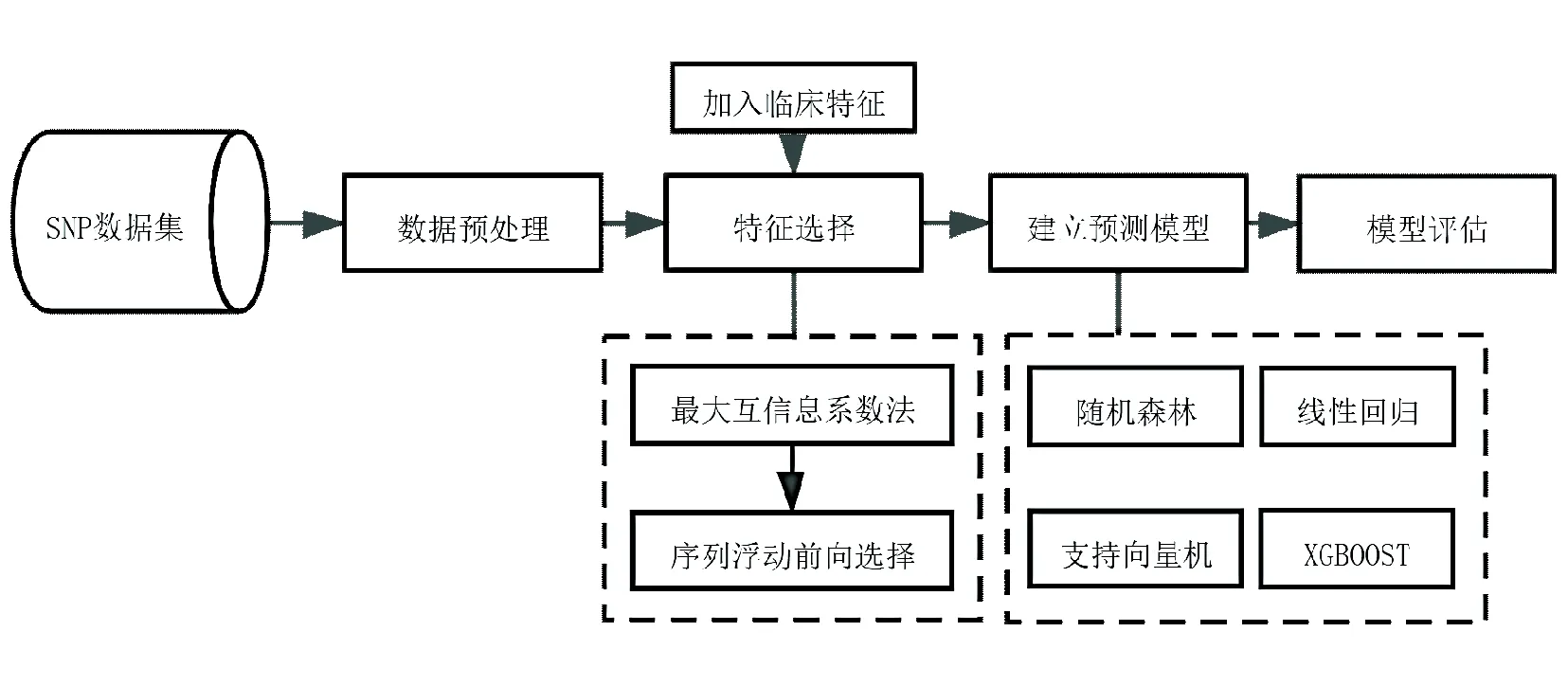
图1 骨密度回归模型分析流程Fig.1 Flow chart of BMD regression model
1.2.1数据预处理:①数据转换:使用Plink全基因组关联分析工具通过加性显性编码将SNP基因分型数据格式(AA, AC, CC, NC)转换成数值形式(0, 1, 2, NC)并存放在文本文件中。②数据补缺:SNP基因分型数据集中缺失的值通常标记为“NC”。数据补缺遵循以下标准:分型后“NC”值多于10 %的SNP位点将被丢弃,其余位点的“NC”值将被该位点的众数(SNP数据集中每个位点出现频率最多的编码)替换。
预处理完成后,SNP数据集包含2 263例样本,35 780个特征。由此,特征组合分为临床风险因素特征组、临床风险因素结合遗传因素特征组。临床风险因素特征组由年龄、性别(男性:1,女性:2)、体重、身高、BMI及其对应项平方组成,共10维特征。在此基础上加入SNP位点特征即为临床风险因素结合遗传因素特征组。
1.2.2特征选择:本文提出一种两阶段特征选择算法,首先以MIC作为过滤式方法剔除SNP数据集中大量的噪声数据,再结合临床特征变量使用序列浮动前向选择算法(sequential floating forward selection, SFFS)这一封装式算法,获得信息最丰富的特征子集。
MIC由 Reshef等[10]提出,它基于互信息度量变量对之间的相关性,在数据量巨大的情况下,互信息能够有效地表述变量间的非线性相关关系[11]。
特征Xi与预测变量Y的MIC定义如下:给定双变量(Xi,Y)组成的数据集D,首先进行网格分区形成维度为(x,y)的网格G。对于给定的D,改变网格G的划分方式,D∣G表示D在G上的概率分布,与落在每个子格内的散点的数量成正比,计算不同划分方式下的最大互信息。定义特征矩阵M(D)x,y=(mx,y),其中mx,y是任意x×y网格所获得的最高归一化互信息值,特征矩阵的第(x,y)项mx,y为:
M(D)x,y=[maxI(D∣G)]/log2min(x,y)
(1)
定义统计值MIC:
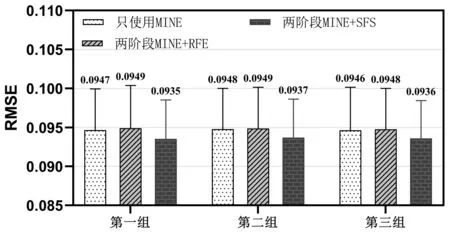
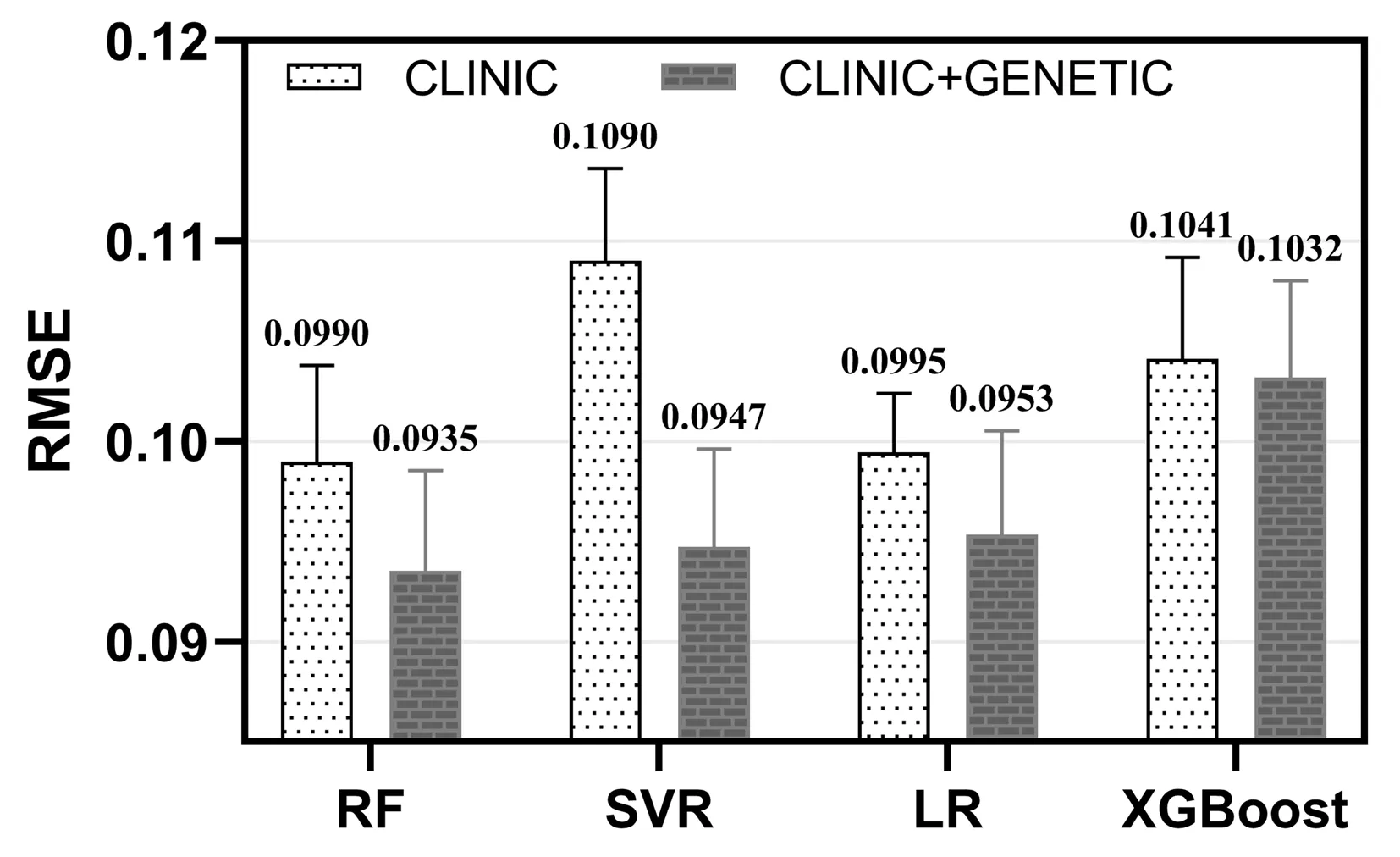
MIC(Xi,Y)=maxM(D)x,y,xy (2) 其中B是关于样本大小的函数,通常设B=n0.6,n为训练集中的样本数。 本文利用MINE(maximal information-based nonparametric exploration)算法[12]计算位点特征与预测变量BMD之间的MIC ,选择MIC得分最高的前m个SNPs作为下一阶段特征选择方法的输入。 SFS算法从一个空的特征子集开始,通过不断添加(或移除)特征直到选择出最优特征子集或达到预先指定的子集大小。SFFS是增加了回退机制的SFS算法。 具体的特征选择流程见图2。为了选择出信息最丰富的特征子集,首先利用MINE算法计算SNP特征与BMD之间的MIC[10],选择MIC得分最高的前m个SNPs结合临床数据作为下一阶段SFFS的输入。特征在SFFS算法中经过k次迭代,在每次迭代过程中,以准则函数最大化为目的,从特征空间中选择一个最佳特征,并通过额外的排除或包含步骤,检查若移除一个特征后,特征子集能否提高预测的性能。最后根据不同数量的特征组合,选择达到最优预测效果的特征子集N。提出的特征选择算法在python 3.7上实现。 1.2.3预测模型:本研究采用随机森林[13]作为主要的预测模型,构建机器学习预测模型的一个关键步骤是优化超参数以获得最佳模型性能,选择优化以下两个随机森林模型的超参数:①n_estimators,森林中决策树的数量。②max_features,建立决策树时选择的最大特征数。首先使用网格搜索确定超参数n_estimators的最优值,然后选择max_features。 通过十折交叉验证确定最终的最优特征子集。除随机森林之外,另有几种经典的回归算法,即支持向量机回归(SVR)、线性回归(LR)、XGBoost用于测试我们提出的两阶段特征选择方法以及最优特征子集的有效性和稳定性。 1.2.4评估方法:为衡量回归模型的预测精度和泛化能力,使用均方根误差为指标对模型进行评估。均方根误差是评估回归模型与数据集拟合程度的一种方法,其计算公式为: (3) 其中,Yi是数据集中第i个样本的预测值,f(xi)是数据集中第i个样本的实际值,N是样本容量。 本研究使用2 263例白种人样本的临床风险因素和遗传因素数据集,对上文提出的基于机器学习的骨质疏松症致病因素分析方法进行验证。 在对数据集进行预处理后,SNP数据集包含35 780个位点特征。首先以MIC作为过滤式方法剔除SNP数据集中大量的噪声数据,最终保留MIC得分最高的前100个SNP作为遗传特征,与临床特征混合进行下一步的SFFS,选择过程基于随机森林回归模型,在样本数据集中进行十折交叉验证,评估标准为RMSE。 如图3所示,当特征数为57时,模型的RMSE达到最低为0.093 598 g/cm3,即这组特征(包含51个SNP位点,6个临床特征)能够最好地拟合实际的骨密度值。 在训练模型之前对随机森林回归模型的超参数进行选择,表2为训练和优化随机森林算法所测试的超参数及其对应的测试值和最终确定的取值。 表2 随机森林模型超参数取值Table 2 Hyperparameters in random forest model 网格搜索发现分类器个数的最优值为170、建立决策树时选择的最大特征数的最优值为25,此时模型的均方根误差最低。 在模型输入为临床因子特征变量、遗传因子加临床因子特征变量两种情况下,以随机森林为回归模型,进行3组实验。测试集的 RMSE值如图4所示,每组实验结果的均值以数字标注在条形图之上,标准差以误差线形式表示。 图4 加入遗传因素前后模型的RMSE值Fig.4 The effects of introducing genetic factors on RMSE value of the model 分析加入遗传因素前后模型的均方根误差,加入遗传因素前,3组十折交叉验证实验的RMSE分别为(9.88±0.51)×10-2g/cm3、(9.90±0.48)×10-2g/cm3、(9.88±0.52)×10-2g/cm3,30次实验的平均RMSE为(9.89±0.51)×10-2g/cm3;加入遗传因素后,3组十折交叉验证实验数据的RMSE分别为(9.35±0.50)×10-2g/cm3、(9.37±0.50)×10-2g/cm3、(9.36±0.49)×10-2g/cm3,30次实验的平均RMSE为(9.36±0.50)×10-2g/cm3。 相比只以临床危险因素为特征,在加入遗传因素后,3组交叉验证中模型的RMSE分别降低了5.36 %、5.35 %、5.26 %,30次实验的平均RMSE降低了5.36 %。说明加入遗传因素作为特征变量能够降低模型的均方根误差,使骨密度回归模型更好地拟合数据集。 为证明如上提出的融合最大互信息系数和序列浮动前向选择这一两阶段特征选择方法的有效性,使用相同的数据集,混合遗传因素与临床因素特征,将本研究所提两阶段特征选择方法与仅使用MINE算法、以及第一阶段使用MINE算法,第二阶段使用另一种经典的封装式算法递归特征消除(recursive feature elimination, RFE)进行对比。 如图5所示,仅使用最大互信息系数进行特征选择,3组十折交叉验证实验数据的RMSE分别为(9.47±0.53)×10-2g/cm3、(9.48±0.52)×10-2g/cm3、(9.46±0.55)×10-2g/cm3,30次实验的平均RMSE为(9.47±0.54)×10-2g/cm3;第一阶段使用最大互信息系数,第二阶段换用递归特征消除(RFE)选择最优特征子集,3组十折交叉验证实验数据的RMSE分别为(9.49±0.55)×10-2g/cm3、(9.49±0.53)×10-2g/cm3、(9.48±0.52)×10-2g/cm3,30次实验的平均RMSE为(9.48±0.54)×10-2g/cm3;本研究所提混合最大互信息系数与序列浮动前向选择的特征选择算法30次实验的平均RMSE为(9.36±0.50)×10-2g/cm3,为3种方法中最低。 图5 所提特征选择方法与仅使用MINE算法、第一阶段MINE算法+第二阶段RFE选择特征的效果对比Fig.5 Comparison of RMSE for MINE, MINE+RFE and the proposed MINE+SFS method 为证明所提两阶段特征选择方法以及遴选出来的特征子集的稳定性,将最优特征子集输入不同的回归模型(RF, SVR, LR, XGBoost)进行十折交叉验证。其中,SVR模型采用的核函数为径向基核函数(RBF),已经对输入特征和标签值进行数据标准化处理。LR及XGBoost采用标准参数。对比结果如图6所示。 图6 特征子集在使用不同回归模型时的RMSEFig.6 Comparison of RMSE for different regressors 加入遗传因素作为特征变量前后,RF的RMSE分别为(9.90±0.48)×10-2g/cm3和(9.35±0.50)×10-2g/cm3;SVR的RMSE分别为(10.90±0.46)×10-2g/cm3和(9.47±0.49)×10-2g/cm3;LR的RMSE分别为(9.95±0.29)×10-2g/cm3和(9.53±0.52)×10-2g/cm3;XGBoost的RMSE分别为(10.41±0.51)×10-2g/cm3和(10.32±0.49)×10-2g/cm3。在使用本文提出的两阶段特征选择算法所遴选出来的特征子集后,几种回归模型的RMSE都有明显降低,其中随机森林回归模型在临床因素混合遗传因素特征集上RMSE最低。 相较于既往的骨质疏松单一致病因素分析研究,本文引入遗传因素,旨在基于SNP数据集结合临床危险因素实现对这一复杂疾病更准确、更鲁棒的预测。为提高算法性能并减少时间复杂度,同时保留特征的生物学含义和解释性,采用最大互信息系数作为过滤式方法剔除SNP数据集中大量的噪声数据,最终保留MIC得分最高的前100个SNP作为遗传特征,与临床特征混合,基于随机森林回归模型进行下一步的序列浮动前向选择。初步构建出具有良好的预测准确度和稳定性的骨质疏松症致病因素分析方法。 研究提出的两阶段特征选择方法兼顾封装式方法的精度和过滤式方法的效率,可以实现时间复杂度较低的非线性预测模型,降低预测误差,为骨质疏松症以及类似的复杂疾病的致病因素探明、预测模型的建立提供有价值的参考。 低骨密度可能是多种致病途径的共同结果,这些途径受到遗传因素的影响,本研究基于2 263例白种人样本,建立骨密度机器学习回归模型,筛选出如表2的51个骨质疏松症易感位点。 表3 方法筛选出的51个易感位点Table 3 Characteristics of 51 SNPs selected by the proposed method 这些位点位于13个基因,其多态性通过多种途径影响骨密度。基因GNG12-AS1、WLS、MEF2C、CDKAL1和SFRP4通过激活Wnt/β-catenin信号通路调控骨形成,Wnt/β-catenin信号通路及相关蛋白在骨细胞分化、增殖和凋亡的过程中至关重要[14-17]。基因SUPT3H与PKDCC调控间充质干细胞与软骨细胞的分化过程[18-19]。AKAP11、RPS6KA5与SMG6参与骨细胞生长发育过程,在此前的研究中被证实与骨质疏松特征具有显著相关性[20-24]。除了基质蛋白和骨细胞的平衡外,骨骼的完整性还依赖于矿物质的稳态。GALNT3调控循环中的磷酸盐水平[17],KCNMA编码细胞上Big K+(BK)大电导钙和电压激活的K+通道的成孔性α亚基[25],参与骨吸收。基因ESR1编码雌激素受体α,在调节骨量和骨质疏松症的发生中发挥重要作用[26]。 值得注意的是,在我们识别出的易感位点中,已经有一部分在此前的GWAS研究被鉴定出与骨质疏松、骨折等性状显著相关,即rs726282[27]、rs932477[28]、rs2179922[29]、rs2941740[30]、rs4952590[31]、rs6721582[32],这进一步证实了本文所提方法的有效性。本文创新性的将临床风险因素和遗传因素结合,通过机器学习方法识别骨质疏松症易感位点。所提分析方法识别出的其他致病SNP揭示了遗传因素之间、遗传因素与临床因素之间相互作用的存在,既有在临床上对骨质疏松症的预测意义,也为我们未来对骨质疏松症致病因素的进一步探究提供了潜在的靶点。 研究存在一定的局限性:其一,数据样本规模较小。后续研究将结合更大样本的数据集对分析方法的性能进行进一步的验证,并在全基因组范围内探索骨质疏松症致病因素,完善分析方法,提高模型的计算效率和识别能力,以期全方位且高效地识别骨质疏松症的关联位点和基因;其二,研究没有对分析方法的运算时间进行定量分析,后续研究可以尝试选择不同数量的特征子集、优化机器学习算法等方法以提升模型的时间性能。2 结果

2.1 加入遗传因素对模型均方根误差的影响

2.2 不同特征选择策略对模型均方根误差的影响
2.3 使用不同回归模型的对比分析
3 讨论

猜你喜欢
分子催化(2022年1期)2022-11-02
中学生数理化(高中版.高一使用)(2021年9期)2021-12-02
安庆师范大学学报(自然科学版)(2021年1期)2021-11-28
烟草科技(2021年6期)2021-06-24
生物学教学(2018年4期)2018-11-29
电脑知识与技术(2018年19期)2018-11-01
现代电子技术(2016年23期)2017-01-12
电脑知识与技术(2016年25期)2016-11-16
电脑知识与技术(2016年15期)2016-07-04
电脑知识与技术(2016年14期)2016-06-30
