
BLim1/2+(m=6、7)团簇的结构及储氢性能的研究
2023-01-09 02:05徐丝雨刘婷婷
井冈山大学学报(自然科学版) 2022年6期
徐丝雨,刘婷婷,康 闽,朱 玮,阮 文
BLim1/2+(m=6、7)团簇的结构及储氢性能的研究
徐丝雨,刘婷婷,康 闽,朱 玮,*阮 文
(井冈山大学数理学院,江西,吉安 343009)
为开发新型储氢材料提供更为丰富的理论基础,采用B3LYP泛函在6-311++G (d, p)基组水平上对BLi6+超碱团簇和BLi72+超碱土团簇的稳定性结构、电荷分布等方面进行理论研究,进而研究团簇的储氢性能。结果表明:两个离子团簇均比它们所对应的中性团簇均具有较高的动力学稳定性。两个离子团簇中的每个Li原子同时有效吸附2个氢分子,BLi6+团簇中氢分子在团簇表面平均吸附能为0.969~2.162 kCal/mol,储氢质量分数达31.56 wt%。而BLi7+团簇中氢分子在团簇表面平均吸附能为1.764~3.714 kCal/mol,储氢质量分数达32.21 wt%。它们的储氢性能表明BLi6+团簇和BLi72+团簇均有望成为良好的储氢媒介。
密度泛函理论;BLi6+超碱团簇;BLi72+超碱土团簇;储氢
0 引言
随着环保意识的加强,清洁高效的能源尤为重要。其中,氢能被人们称之为21世纪的“终极能源”。但是为了更好应用氢能,必须进一步完善氢的规模制备、储存和运输,而氢的储存是关键的技术难题[1]。在各种方法中,利用储氢材料储氢不仅能量密度高而且安全性好,被誉为最具发展前景的储氢方法。由于碳或硼基纳米材料重量轻,比表面积高,有利于氢分子的吸附,所以金属原子修饰碳或硼基纳米材料成为了储氢材料的典型代表。例如Zhao等[2]研究了Sc修饰硼富勒烯,储氢质量分数达8.77 wt%。虽然纳米材料由金属原子修饰后具有更优的吸附储氢本领,但是,在储氢的同时,纳米材料表面的多个金属原子趋向形成簇,并且这种聚合对材料的循环储氢不利[3]。不过,由于碱金属和碱土金属原子的内聚力相对而言比较小,因此利用它们来修饰此类材料,可以有效地避免聚合[4]。
有一类分子被称为超碱金属,它们的电离势由于其原子间的聚集效应,因此比碱金属更低[5]。低电离势的性质使得超碱金属的最外层价电子易于形成低束缚电子。李莹等[6]指出“超原子”在与其它分子发生反应时,能够保持自身的完整性,因此,它们是设计新型纳米材料的理想基元。在吸附氢分子时,其吸附能介于弱物理吸附和强化学吸附之间,这种配位化合物中的金属原子主要作用是吸附氢分子,而碳或硼基则作为“装饰”金属原子的宿主[7]。在李莹[8]过去的工作中,证实了BLi6和BLi7分别是一种超碱金属和一种超碱土金属。Pan等[9]发现了一些超碱金属团簇能够吸氢,这表明超碱团簇极可能成为有效储氢材料。基于此,同时我们设想超碱土团簇同样能够有效储氢。因此,在研究BLi6+及BLi72+团簇的物理化学性质的基础上,进一步去分析超碱金属团簇与超碱土金属团簇之间储氢性能的强弱,为开发新型储氢材料提供更为丰富的理论基础。
1 计算方法
对于各类团簇的结构及储氢性能,密度泛函理论的研究已达到非常精准的程度[10]。首先,采用B3LYP泛函在3-21G基组水平上对BLim(m=6、7)、BLim1/2+(m=6、7)团簇进行结构优化,其中BLi6和BLi6+均为正八面体型团簇、BLi7和BLi7+均为五角双锥型团簇。初步得到它们的稳定结构后,在6-311++G (d, p)基组水平上进一步进行优化,从而得到它们的最稳定结构(如图1)。最后,利用同种方法来构建氢分子被吸附在团簇表面的稳定结构(如图3 -图4),并研究它们的物理化学性质。分析过程中,优化结构的稳定性根据振动频率来判断,利用频率计算来分析优化结果是极小值还是鞍点。如果体系中存在虚频,则采用消虚频程序来进行适当的调整,以保证体系处于势能面中的局域最小点,从而得到真正的最稳定结构。本文的所有计算利用Gaussian 09程序[11]完成。

图1 BLi6+(a)和BLi72+ (b)团簇的稳定结构

图2 BLi6+·nH2结构模型(n=6,12)
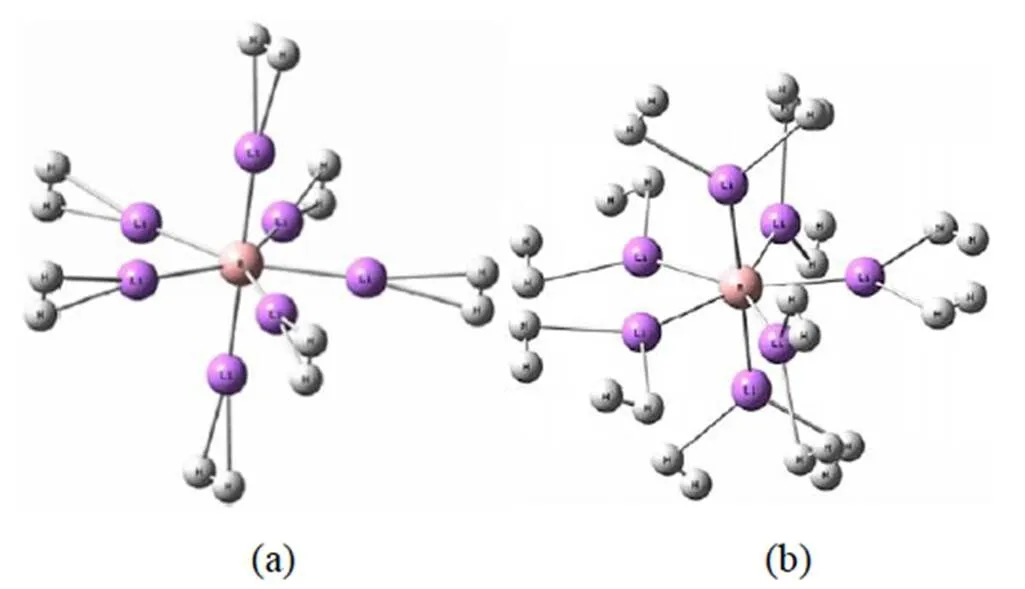
图3 BLi72+·nH2结构模型(n=7,14)
BLi6、BLi6+、BLi7、BLi72+团簇能隙(g) 和平均结合能(b)的计算公式:
g=(LUMO)-(HOMO) (1)
b=[(B)+6(Li)-(BLi6)]/7 (2)
b=[(B)+7(Li)-(BLi7)]/8 (3)
b=[(B5-)+6(Li+)-(BLi6+)]/7 (4)
b=[(B5-)+7(Li+)-(BLi72+)]/8 (5)
上式中,(LUMO)代表团簇的最低空轨道电子能量,(HOMO)代表团簇的最高占据轨道电子能量;(B)、(B5-)、(Li)、(Li+)、(BLi6)、(BLi6+)、(BLi7)、(BLi72+)分别代表B原子、B5-离子、Li原子、Li+离子、BLi6团簇、BLi6+离子团簇、BLi7团簇、BLi72+离子团簇的基态能量。
BLi6+·nH2和BLi72+·nH2中H2的平均吸附能(ad) 和连续吸附能(ac)的计算公式:
ad=[(BLi6+)+n(H2)-(BLi6+·nH2)]/n (6)
ad=[(BLi72+)+n(H2)-(BLi72+·nH2)]/n (7)
ac={[BLi6+·(n-6)H2]+6(H2)-(BLi6+·nH2)}/6 (8)
ac={[BLi72+·(n-7)H2]+7(H2)-(BLi72+·nH2)}/7 (9)
上式中,(H2)、(BLi6+·nH2)、E(BLi72+·nH2)分别代表H2、BLi6+·nH2、BLi72+·nH2团簇的基态能量,n为氢分子数。
BLi6+·nH2稳定体系的储氢质量分数的有关计算公式为:

BLi72+·nH2稳定体系的储氢质量分数的有关计算公式为:

式中,(H)、(Li)、(B)分别为氢、锂、硼原子的相对原子质量。
2 结果与讨论
2.1 BLi6和BLi6+的几何构型和电子性质
在研究BLi6+团簇的结构及储氢性能的同时,对BLi6团簇也进行计算。通过对BLi6和BLi6+团簇的几何结构进行优化,得到虚频为零的最稳定结构,其对称性均为Oh群的正八面体(如图1),且由表1可知,基态BLi6团簇的B-Li键长为2.115 Å,这与李莹[8]采用四种方法优化得到对称性结构吻合,且B-Li键长分别为2.124 Å、2.167 Å、2.177 Å、2.191 Å,与我们的计算结果同样吻合。而BLi6+团簇与李莹等[6]在MP2/6-311 +G(d) 基组水平下计算下结果仍是一致。通过计算可知BLi6+团簇的B-Li键长比BLi6团簇伸长了0.52%,但是BLi6+团簇的结合能b大约是BLi6的8倍之大,这表明虽然BLi6+团簇较BLi6团簇结构略微松散,但是其原子间的结合强很多,足以忽略这种相对的松散。而且,由表1可知,BLi6团簇的前线轨道能隙为1.799 eV,而BLi6+团簇的前线轨道能隙达2.424 eV,增大了34.74%。占据轨道与非占据轨道之间的大前线轨道间隙表明具有高动力学稳定性,这是因为激发能越大,从低分子轨道提取电子并将其添加到高分子轨道中越不易[12]。这表明从电子性质角度出发,BLi6+团簇的动力学稳定性比BLi6团簇的更高。基于此可知,BLi6+属于十分稳定的金属阳离子团簇。进一步对自然电荷进行分析,由表1可知该离子与中性团簇中的中心B原子电荷相差不大,而BLi6团簇上的Li原子的平均电荷为0.565 e,BLi6+团簇上的为0.730 e,这表明BLi6团簇在失去一个电子后,碱金属配体原子上的正电荷明显增多,而电性的增强正表明在空间中形成的静电场更强。根据极化的性质,当附近存在氢分子时,较强的静电场更利于团簇与氢分子的相互作用,从而易于吸氢。因此,BLi6+团簇可作为一个稳定的基底,并通过其来研究对氢分子的吸附行为。

表1 BLi6和BLi6+团簇的结构特征及电子性质
2.2 BLi7和BLi72+的几何构型和电子性质
理论计算BLi72+和BLi7团簇体系,研究可知基态时它们均是对称性为CS群的五角双锥型。其结构特征及电子性质参量列于表4,从表4可知,中性团簇平面内五个几乎等长的B-Li键长为2.235Å,另外两个轴向较短的B-Li键长为2.126 Å。而在离子团簇中,平面内B-Li键长为2.291 Å,另外两个轴向的为2.199 Å。通过计算可知,团簇平面内BLi72+团簇的B-Li键长比BLi7团簇伸长了2.5%,轴向的B-Li键长伸长了3.4%。但是离子团簇的结合能b大约是中性团簇的7倍之大,而且BLi72+团簇的前线轨道能隙比BLi7团簇增大了54.78%,这表明虽然BLi72+团簇比BLi7团簇在结构上更为松散一点,但是从电子性质角度出发,BLi72+团簇比BLi7团簇的动力学稳定性高很多。基于此,BLi72+团簇是一个稳定的碱土金属阳离子。再进一步对电荷发布进行分析,表1中BLi7团簇上的中心B原子上的电荷为-3.618 e,BLi72+团簇上的中心B原子上的电荷为-3.480 e,中心B原子上所带的负电荷相差不大。但是,BLi7团簇平面上的Li原子上的电荷为0.485 e,轴向上的Li原子上的电荷为0.596 e。而BLi72+团簇平面上的Li原子上的电荷为0.776 e,轴向上的Li原子上的电荷为0.799 e。首先,轴向上Li原子的正电荷数比平面上的大,说明轴向上B-Li之间的静电作用力更大,这正也符合俩团簇的轴向上的B-Li键比平面上的更短。其次,当BLi7团簇失去两个电子时,BLi72+中的Li原子上的正电荷明显增多,而B-Li键长的涨幅不大,电性的明显增强说明离子团簇比中性团簇在空间中形成的静电场强很多。当团簇周围存在氢分子时,与BLi7团簇相比,拥有较强静电场的BLi72+团簇更有利于氢分子被极化,加大与氢分子的相互作用,从而有助于氢分子被吸附在离子团簇的表面,所以BLi72+团簇同样可以作为一个稳定的基底来研究其对氢分子的吸附行为。与BLi6+团簇相比,BLi72+团簇可以看做一个Li离子插入BLi6+团簇的Li4平面内,且导致其平面内的B-Li键比BLi6+团簇的长了0.176 Å,轴向的B-Li键长了0.084 Å,可见BLi6+团簇比BLi72+团簇更紧凑,但同时BLi72+团簇展开储氢面积更大,因此从此方面难以判断储氢性能的强弱。

表2 BLi7和BLi72+团簇的结构特征及电子性质
2.3 BLi6+和BLi72+的储氢性能
为进一步分析BLi6+和BLi72+团簇的结构及储氢性能,分别令团簇的每个Li原子附近从一开始采取递增的形式吸附相同数目的氢分子。结果表明,两团簇中每个Li原子附近均可有效吸附2个氢分子 (如图2和图3),分别将BLi6+·nH2和BLi72+·nH2的构型、吸附能以及电荷分布等相关数据列于表3和表4。在相同的计算方法下,孤立氢分子键长为0.744 Å,由表3可知,优化结构中的氢分子键长仅变长0.005 Å,又由表4可知,优化结构中大多数H-H键长处于0.750 ~ 0.795 Å的范围内,均并未发生断键行为,这说明氢分子被Li原子吸附时仍保持分子形态,即BLi6+和BLi72+团簇皆可有效吸附氢分子。氢分子键长的伸长是吸氢中不可避免的,这是由于氢分子与团簇的相互作用,其部分电荷会转移到B原子或者Li原子中,由此同孤立氢分子相比,其间的相互作用会有所减弱。
首先,从键长角度进行分析,由表3可知,在每个Li原子从吸附1个氢分子到2个氢分子的过程中,Li原子与氢分子之间的平均距离增大2.711%,而B-Li的平均键长仅变长0.471%,又由表4可知,该过程中Li原子与氢分子的平均距离增大5.877%,而B-Li平均键长仅变长0.310%,即两团簇的主体仍然保持着原有的结构,这表明BLi6+和BLi72+团簇在吸氢后结构依然具有较高的稳定性。在这一过程中,氢分子的平均吸附能和连续吸附能均减小。结合李莹等[8]。表明中心原子与配体碱金属原子之间的键长越短,并且它的电负性越高,则它们之间的相互作用越强的说法,这说明储氢量越多,相互结合越弱。当每个Li原子周围吸附3个氢分子时,Li原子周围均存在1个氢分子远离Li原子的情况,这说明团簇吸氢已达到极限。
接下来,从吸附能角度进行分析,由表3和表4可知,氢分子的平均吸附能和连续吸附能均随着团簇吸氢数目的增多而呈现减小的状态。其中,当BLi6+和BLi72+团簇的每个Li原子周围分别吸附达2个氢分子时,氢分子的连续吸附能已达到比较小的负值,分别仅为-0.224 kCal/mol和-0.179 kCal/mol,而此处的连续吸附能所反映的正是体系对下一个氢分子的吸附能力,当其为很小的正值或者负值的时候,此体系已经不具备进一步吸附氢分子的能力,因此,这同样可以表明团簇吸氢已达到极限。与BLi6+团簇相比,当每个Li原子吸附相同数量氢分子时,BLi72+团簇的平均吸附能和连续吸附能均更大,尤其是平均吸附能表明了体系的吸氢能力,因此,从吸附能角度可以初步猜测BLi72+团簇的储氢性能更佳。
最后,从体系的电荷分布上来拓展分析BLi6+和BLi72+团簇无法进一步吸附氢分子现象的原因。由上述分析可知两团簇体系在周围空间中存在较强的静电场,导致靠近团簇的氢分子发生电极化现象,氢分子在这种相互作用下而被吸附在团簇表面。然而随着所吸附的氢分子数目的加大,氢分子上的电子密度会重新发布。在以BLi6+团簇为基底的体系中靠近Li原子一侧的密度减小,而远离Li原子一侧的密度增大,不过总体看来,各个氢分子上面的正电子平均有少量减少,Li原子上的正电荷也有少量减少,B原子上的负电荷数同样有所减少,使得BLi6+团簇产生的静电场减弱,从而其与氢分子之间的相互作用减小。当BLi6+团簇中每个Li原子吸附的氢分子达到3个时,团簇与氢分子之间的静电相互作用不能维持氢分子均被吸附,并且较多的氢分子之间会产生较大的库仑排斥作用。与BLi6+团簇所不同的是,BLi72+团簇体系在吸附2个氢分子时,靠近团簇的氢原子带较少负电荷,同一个氢分子中远离团簇的氢原子带稍大的正电荷。取平均后,各个氢分子上面的正电子有少量减少,锂原子上的正电荷却有少量增加,B原子上的负电荷数同样也有所增加,说明BLi72+团簇在吸附2个氢分子时已达到临界点,且与BLi6+团簇相比,在每个Li原子吸附2个氢分子时,BLi72+团簇主体更加稳定,氢分子与Li原子的距离变化较小,可以预测BLi72+团簇比BLi6+团簇吸氢更加稳定。除此以外,由于立体效应,两团簇在吸附3个氢分子时每个Li原子与3个氢分子中的1个氢分子间距突然巨增。因此,BLi6+稳定结构储氢质量分数为31.57wt%,BLi72+稳定结构有效储氢质量分数为32.21wt%,比BLi6+团簇稳定结构的有效储氢质量分数稍大。

表3 BLi6+·nH2团簇结构、吸附能和原子电荷平均分布

表4 BLi72+·nH2团簇结构、吸附能和原子电荷平均分布
3 结语
采用B3LYP泛函在6-311+G(d, p)基组水平上对BLim1/2+(m=6、7)团簇进行结构优化,研究它们的稳定结构和储氢性能。结果表明:碱金属团簇BLi6+与碱土金属团簇BLi72+皆具备良好的储氢性能,且本身的动力学稳定性较高。具体分析如下:硼基由Li原子修饰后,具有较好的吸氢能力。此类团簇储氢是氢分子在与团簇之间发生的静电极化作用下,被吸附在团簇体系周围。以BLi6+团簇的稳定结构作为基底时,氢分子的平均吸附能在0.929~2.126 kCal/mol的范围内变化,而以BLi72+团簇的稳定结构作为基底时,氢分子的平均吸附能在1.767~3.714 kCal/mol的范围内变化,后者的平均吸附能最大和最小值均大于前者,表明BLI72+比BLi6+吸氢能力更强。通过计算可得BLi6+团簇的最大储氢质量分数达31.57 wt%,而BLi72+团簇的最大储氢质量分数达32.21wt%,显然,BLi72+团簇的储氢的最大质量分数更大,且通过对平均吸附能进行比较,BLi72+团簇吸氢更加稳定。BLim1/2+(m=6、7)团簇较高的储氢比例暗示着它们均具有成为储氢材料的潜能。
[1] 刘鑫.物理吸附储氢的最佳条件分析[D].兰州:西北师范大学,2015.
[2] Zhao Y F, Kim Y H,Dillon A C,et al. Hydrogen storage in novel organometallic buckyballs [J]. Phys. Rev. Lett. , 2005, 94: 155504.
[3] Sun Q,Wang Q,Jena P,et al. Clustering of Ti on a C60surface and its effect on hydrogen storage [J]. Am Chem Soc,2005,127:14582.
[4] 阮文,宋红莲,伍冬兰,等. 平面星形Li6Si6团簇的结构及其储氢性能研究[J]. 原子与分子物理学报, 2018, 35(1):68-72.
[5] 肖文敏,麻娜娜,马腾颖,等.超碱金属复合物Al7X0/-(X=F,Cl,Br,I)的结构及非线性光学性质的理论研究[J].高等学校化学学报,2013,34(9):2184-2190.
[6] 李莹,佟晶,吴迪.超碱金属阳离子BM6+,CM5+,NM4+, OM3+和FM2+(M=Li, Na)的理论研究[J]. 分子科学学报, 2010, 26(4):252-255.
[7] Wang Y S, Wang F, Li M, et al. Theoretical prediction of hydrogen storage on Li decorated planar boron sheets[J]. Appl. Surf. Sci., 2012, 258: 8874.
[8] 李莹.分子及团簇的键、结构和性质的理论研究[D].长春:吉林大学, 2008.
[9] Pan S, Merino G, Chattaraj P K. The hydrogen trapping potential of some Li-doped star-like and super-alkali systems [J]. Phys. Chem. Chem. Phys., 2012, 14:10345.
[10] 阮文,冯五强,温在国,等. GeLi62+团簇的结构及储氢性能研究[J].井冈山大学学报:自然科学版, 2019, 40(1): 5-8,18.
[11] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian09, Revision A. 02. Wallingford CT: Gaussian, Inc[S]. 2009.
[12] Tang C M, Wang Z G, Zhang X, et al. The hydrogen storage properties of Na decorated small boron cluster B6Na8[J]. Chem. Phys. Lett., 2016, 661:161
STRUCTURE AND HYDROGEN STORAGE PROPERTIES OF BLim1/2+(m=6, 7) CLUSTERS
XU Si-Yu, LIU Ting-ting , KANG Min, ZHU Wei,*RUAN Wen
(School of Mathematics and Physics, Jinggangshan University, Ji’an, Jiangxi 343009, China)
In order to provide an abundant theoretical basis for the development of new hydrogen storage materials, in this paper the B3LYP method of density functional theory was used to study the stable structure, charge distribution and other properties of BLi6+super alkaline ion clusters and BLi72+super alkaline earth ion clusters at the basis set level of 6-311++G (d, p), so as to study the hydrogen storage performance of clusters. The results show that two ion clusters have higher stability than their corresponding neutral clusters. Every lithium atom in the two ion clusters can effectively adsorb two hydrogen molecules at the same time, the average energy of adsorbed hydrogen molecules on the BLi6+cluster surface is 0.969~2.162 kCal/mol, and the maximum hydrogen storage mass fraction is 31.57 wt%. And the average energy of adsorbed hydrogen molecules on the BLi72+cluster surface is 1.764~3.714 kCal/mol, and the maximum hydrogen storage mass fraction is 32.21 wt%. The hydrogen storage properties of BLim1/2+cluster indicate that BLim1/2+clusters are expected to be a good hydrogen storage medium.
density functional theory (DFT); BLi6+super alkali cluster; BLi72+super alkaline earth clusters; hydrogen storage
O641/O561
A
10.3969/j.issn.1674-8085.2022.06.003
1674-8085(2022)06-0013-06
2022-05-19;
2022-06-25
国家自然科学基金项目( 11764022);江西省自然科学基金项目(20171BAB201020);江西省教育厅科技计划项目(GJJ190559);江西省大学生创新创业训练计划项目(S202010419042)
徐丝雨(1999-),女,江西九江人,井冈山大学数理学院物理专业2018级本科生(Email:2548350203@qq.com);
*阮 文(1970-),男,江西吉安人,教授,博士,主要从事纳米材料和团簇物理研究(E-mail:9919950043@jgsu.edu.cn).
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
电力科技与环保(2022年3期)2022-07-15
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
无机盐工业(2022年3期)2022-03-11
青岛大学学报(工程技术版)(2019年2期)2019-09-10
汽车实用技术(2018年7期)2018-05-18
中学化学(2015年8期)2015-12-29
中学化学(2015年5期)2015-07-13
中学化学(2015年5期)2015-07-13
