
基于DFT 密度泛函理论的聚酯热降解反应机理研究
2023-01-16 02:54张家龙关震宇朱有财
分子催化 2022年5期
张家龙, 关震宇, 朱有财, 刘 振*
(1. 中国石油化工股份有限公司 上海石油化工研究院, 上海 201208; 2. 华东理工大学 化工学院, 上海 200237)
聚酯(Polyester)是由多元酸与多元醇经酯化反应和缩聚反应生成的一类高分子聚合物的统称[1].聚对苯二甲酸乙二醇酯(Polyethylene Terephthalate,简称PET)是最为常见的一类聚酯高分子材料. 由于PET具有优良的物理、 化学以及机械性能等特点,被广泛应用于纤维、 容器、 薄膜、 瓶用、 胶片、 塑料等领域, 目前已成为我国聚酯产品中产量最大、 品种最多的一类合成材料[2].
通过缩聚反应生成的PET长链, 在反应釜中会发生热降解副反应, 在后续的纺丝、 挤压或成型过程中亦会发生热降解, 进而引起PET聚酯分子量降低及PET聚酯变色[3]. 研究表明, PET链的热降解主要是由分子内酯键发生β链裂解反应导致, 该反应会生成具有乙烯基酯和端羧基的热降解产物,其反应机理如图1所示[4]. Buxbaum等[5]使用模型化合物详细研究了PET热降解反应机理, 结果表明PET通过β链裂解反应发生热降解, 并没有证据表明热降解过程中存在自由基. Tomita等[6]将二苯甲酸乙二酯(EDB)作为PET的模型化合物, 研究发现EDB的热降解速率遵循一阶动力学, 其活化能Ea为223.4 kJ/mol. Hergenrother等[7]利用红外光谱法测得的PET热降解活化能Ea为197.9 kJ/mol.
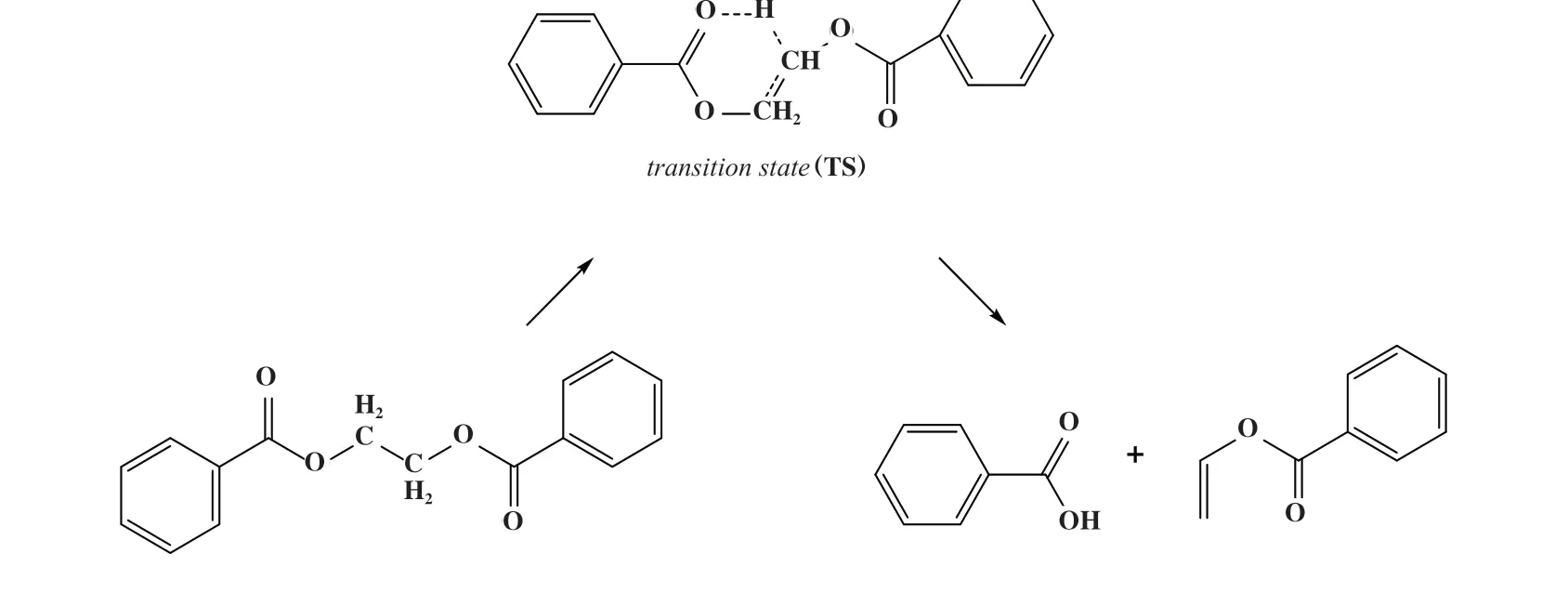
图1 PET链内酯键的β 消除反应机理示意图Fig.1 Reaction scheme of β-elimination reaction at ester linkage of PET
催化剂对PET热降解β消除反应存在一定的作用. PET工业生产中最常用的缩聚催化剂是锑系催化剂. 锑系催化剂主要品种有氧化锑、 醋酸锑和乙二醇锑. 虽然锑催化剂在催化活性、 颜色和成本方面有很好的平衡, 但是锑系催化剂对人体有损害,并且在后道工序中污染环境[8-11]. 钛金属是一种资源丰富, 无毒的环保型金属, 并且以乙二醇钛为代表的钛系聚酯催化剂具有较高的催化活性, 但其得到的聚酯产品色相不佳, 略带黄色[3]. 有研究报道认为钛系催化反应体系中会发生以聚酯热降解为主的副反应, 但关于钛系催化剂聚酯产品黄变的机制尚不明确[10-11].
近年来, DFT方法被用来研究聚酯缩聚及热降解的反应机理[12-13]. 在现有基础上, 我们采用DFT方法对钛系催化剂在聚酯热降解反应中的作用进行理论研究, 通过解析参与反应的催化剂分子结构和反应能量, 旨在阐明催化剂作用机制及聚酯热降解的反应机理, 为工业生产中通过改进钛系催化剂体系来抑制聚酯热降解副反应的发生提供理论指导.
1 计算方法与模型
计算工作如结构优化、 频率计算以及内禀反应坐标IRC(Intrinsic Reaction Coordinate)验证都在高斯(GAUSSIAN)程序包中完成. 我们不设置任何对称性限制, 采用非局域泛函B3LYP, 对所有原子使用def2-SVP基组进行结构优化. 所有的过渡态结构都经过内禀反应坐标IRC进行验证, 以确认过渡态连接相关反应物和产物. 在几何优化中, 溶剂化效应采用了SMD溶剂模型, 色散校正则采用Grimme等开发的DFT-D3(zero-damping)方法. 根据实验反应条件, 基于B3LYP-GD3/def2-SVP(SMD)理论水平下优化的结构, 在0.1 MPa、 250 ℃条件下进行热力学分析, 并得到相应的Gibbs自由能校正项. 最后,在B3LYP/def2-TZVP理论水平下计算相应的高精度电子能. 因此, 文中报道的所有能量均是经过高精度电子能校正和Gibbs自由能校正后的能量. 在分子模型方面, 采用EDB作为PET的模型分子. 分子静电势(MESP)、 最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)均在Multiwfn软件包中计算, 并使用VMD程序绘图.
2 结果与讨论
2.1 催化剂分子模型
考察了钛乙氧基配合物Ti(OEt)4及其阳离子Ti-(OEt)3+两种催化剂对EDB热降解反应的影响, 其几何优化后的分子结构如图2所示. 钛乙氧基配合物Ti(OEt)4为四配位的八面体结构, 而具有阳离子中心的Ti(OEt)3+催化剂则是三配位的四面体构型. 两种催化剂的C-O与C-C键长相近, 但Ti-O的键长分别为0.183 和0.177 nm.
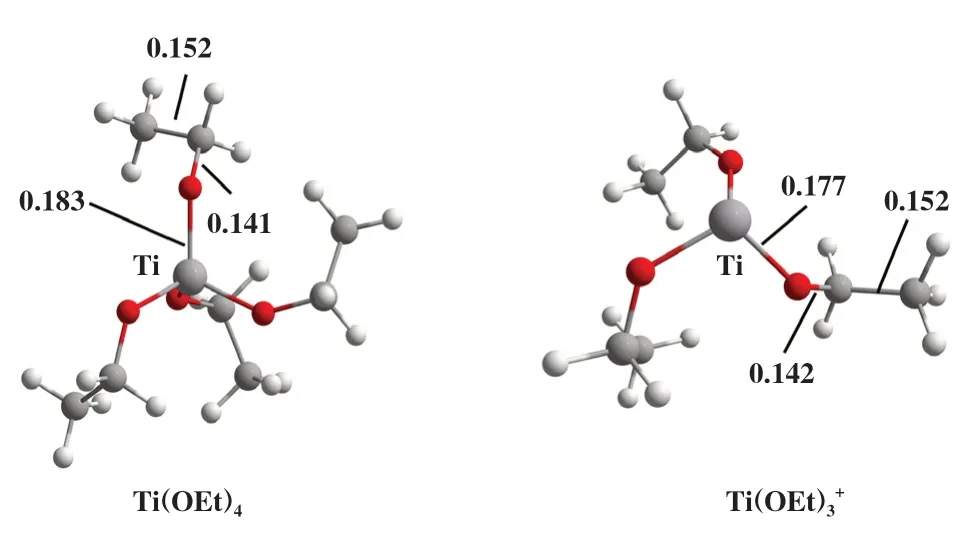
图2 催化剂模型Fig.2 Catalyst models
2.2 无催化剂下的聚酯热降解反应
在无催化剂条件下, EDB发生β裂解的反应路径如图1所示. 其反应物、 过渡态及产物的几何结构如图3所示.
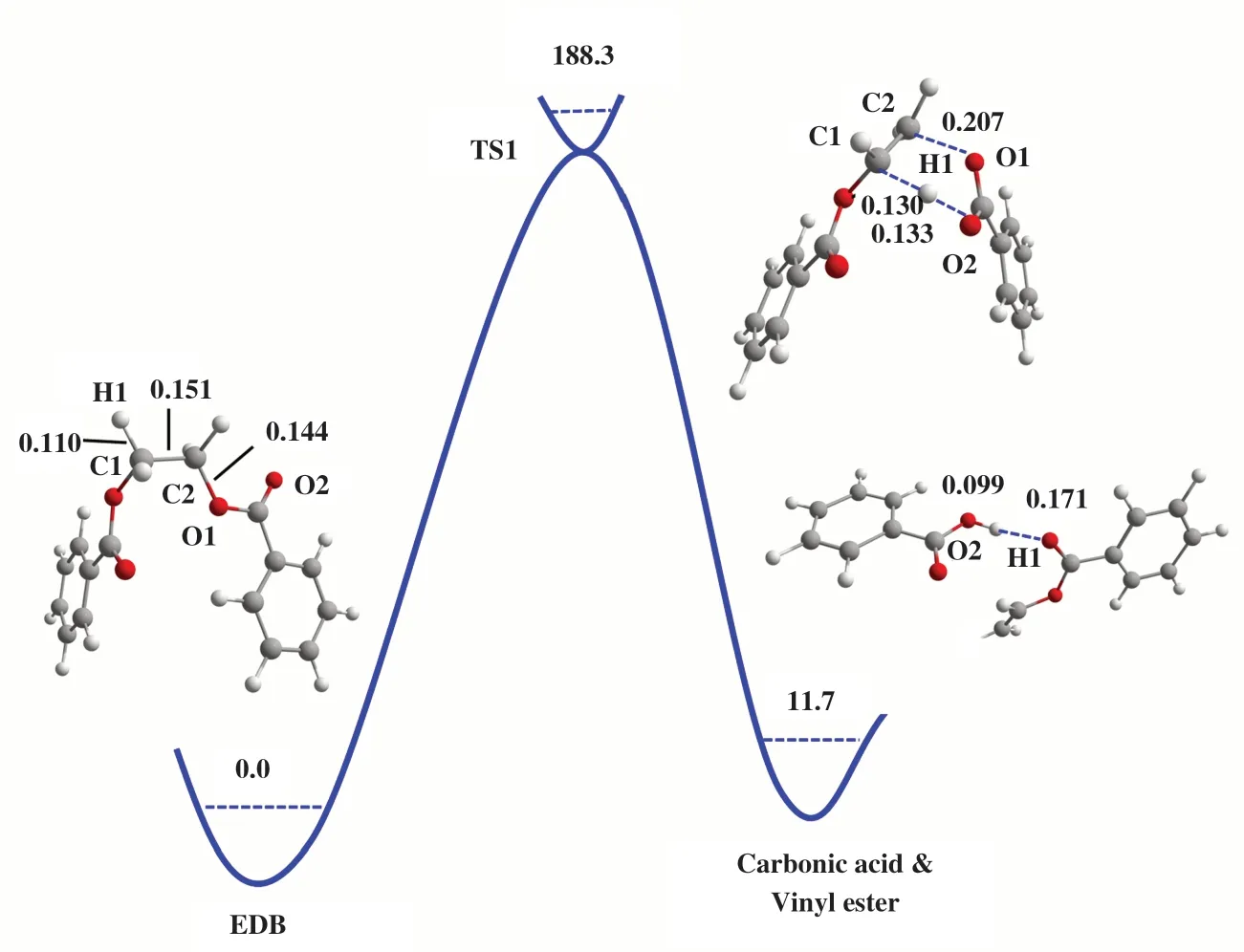
图3 无催化剂下EDB β 消除反应路径中反应物、 过渡态和产物的优化结构及相对能量(kJ/mol)键距以nm为单位Fig.3 Gibbs free energies (kJ/mol) and optimized geometries of the reactants (left), TS (top) and product (right) complexes in the β elimination reaction of EDB without catalyst Bond distances are in nanometers
在该反应中, 酯基的羰基氧原子作为亲核试剂攻击乙二醇单元的β氢原子, 从而形成六中心的环状过渡态. 随后β氢原子转移至酯基的羰基氧原子, 链内的C-O键断裂, 形成产物苯甲酸和苯甲酸乙烯酯. 过渡态TS1 的C2-O1 的键长由反应物的0.144延长至0.207 nm, H1-C1的键长由0.110延长至0.130 nm. 在无催化剂作用下, EDB分子内β消除反应能垒为188.3 kJ/mol, 该数值与Hergenrother等测得的PET热降解活化能(197.9 kJ/mol)相近[7].同时, 产物比反应物能量高11.7 kJ/mol, 表明在无催化条件下生成苯甲酸和苯甲酸乙烯酯在热力学和动力学上均不利于热降解反应的自发进行.
为了理解无催化剂条件下EDB难以发生β裂解的内在机制, 我们对EDB分子进行了静电势和前线轨道分析. EDB分子模型的优化结构, 分子静电势和HOMO、 LUMO以及HOMO-LUMO的能隙如图4所示.
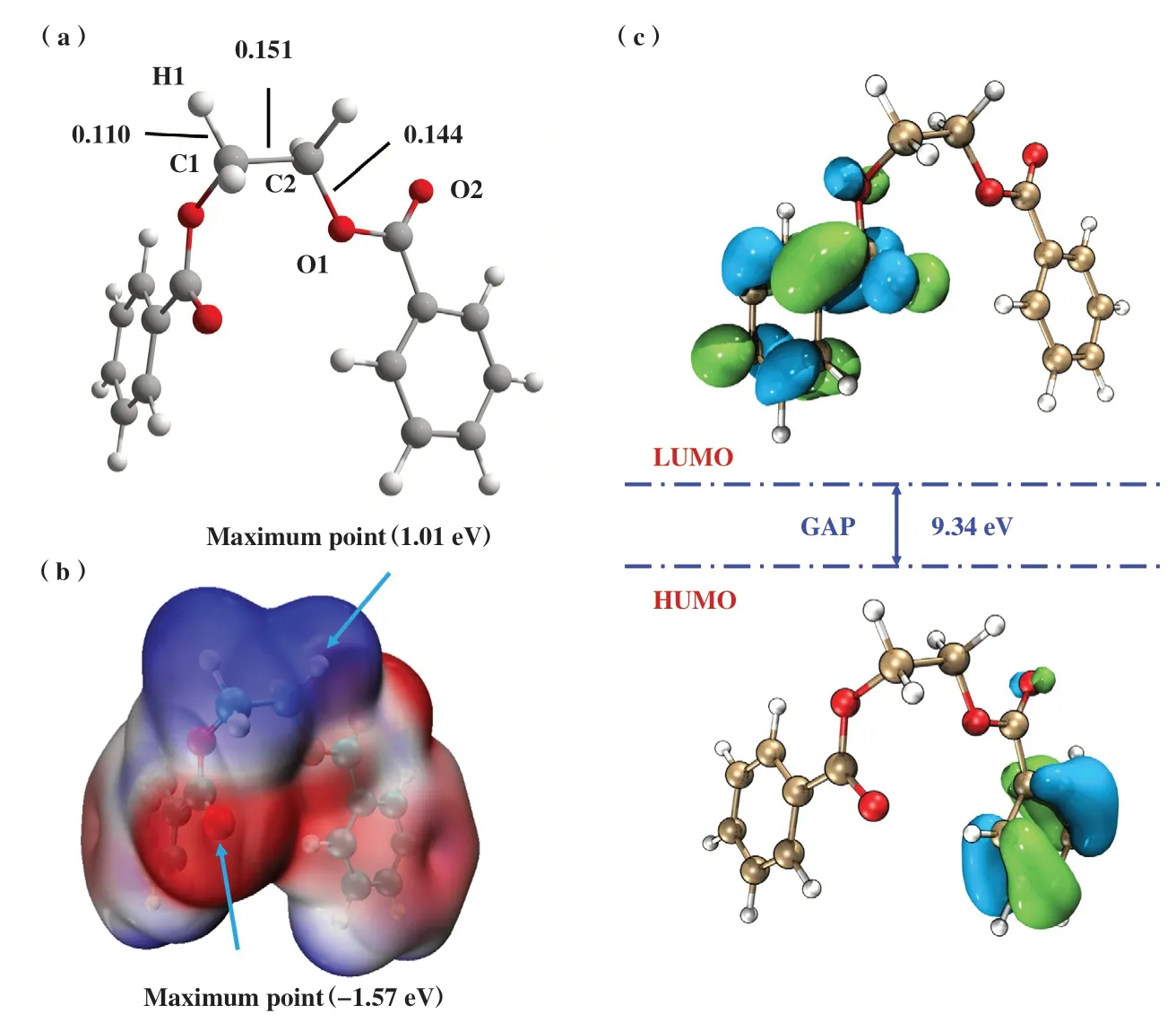
图4 EDB模型(a) 结构优化; (b) 分子静电势; (c) HOMO、 LUMO以及HOMO-LUMO能隙Fig.4 Model of EDB(a) Optimized structure; (b) Electrostatic potential surface map; (c) HOMO, LUMO and HOMO-LUMO energy gap
优化后EDB分子结构中的3 个关键键长参数C1-C2、 C1-H1 和C2-O1 分别为0.151、 0.110 和0.144 nm(图4(a)). 通过对EDB分子结构静电势的分析, 可以直观判断EDB分子的反应位点(图4(b)). 分子静电势(MESP)的正值以蓝色表示, 负值区域以红色表示. 具有孤对电子的羰基氧为分子静电势的最小值(-1.57 eV), 而β-H原子具有分子静电势的最大值(1.01 eV), 在降解反应中β-H倾向于向羰基氧发生转移(β裂解). 从图4(c)可以看出, 反应物EDB的HOMO、 LUMO分别位于聚酯两侧的苯环, 并且HOMO-LUMO能隙高达9.34 eV, 因此, EDB在无催化剂的条件下较难进行分子内的酯键断裂反应.
2.3 Ti(OEt)4催化聚酯热降解反应机理
在Lewis酸催化聚酯热降解反应机理(M1机理)中, 产物的形成分两步进行: (i) 反应物EDB的羰基氧配位至金属中心, 如图5所示, EDB与Ti(OEt)4催化的结合方式为弱配位, Ti-O键长为0.365 nm. (ii)酯基的羰基氧原子作为亲核试剂进攻乙二醇单元的β-H原子, 从而形成六中心的环状过渡态, 随后β氢原子转移到酯基的羰基氧原子, 链内的C-O键断裂, 形成产物苯甲酸和苯甲酸乙烯酯. 与无催化剂热降解过程不同的是, 该产物比反应物能量低38.1 kJ/mol, 表明在Ti(OEt)4催化剂存在条件下生成苯甲酸和苯甲酸乙烯酯在热力学上有利于反应正向进行. 但该过程需要克服的能垒为184.1 kJ/mol, 与无催化剂时的β消除反应能垒(188.3 kJ/mol)相近,说明在Lewis酸催化聚酯热降解反应机理中Ti(OEt)4对热降解反应并未表现出显著的促进作用.
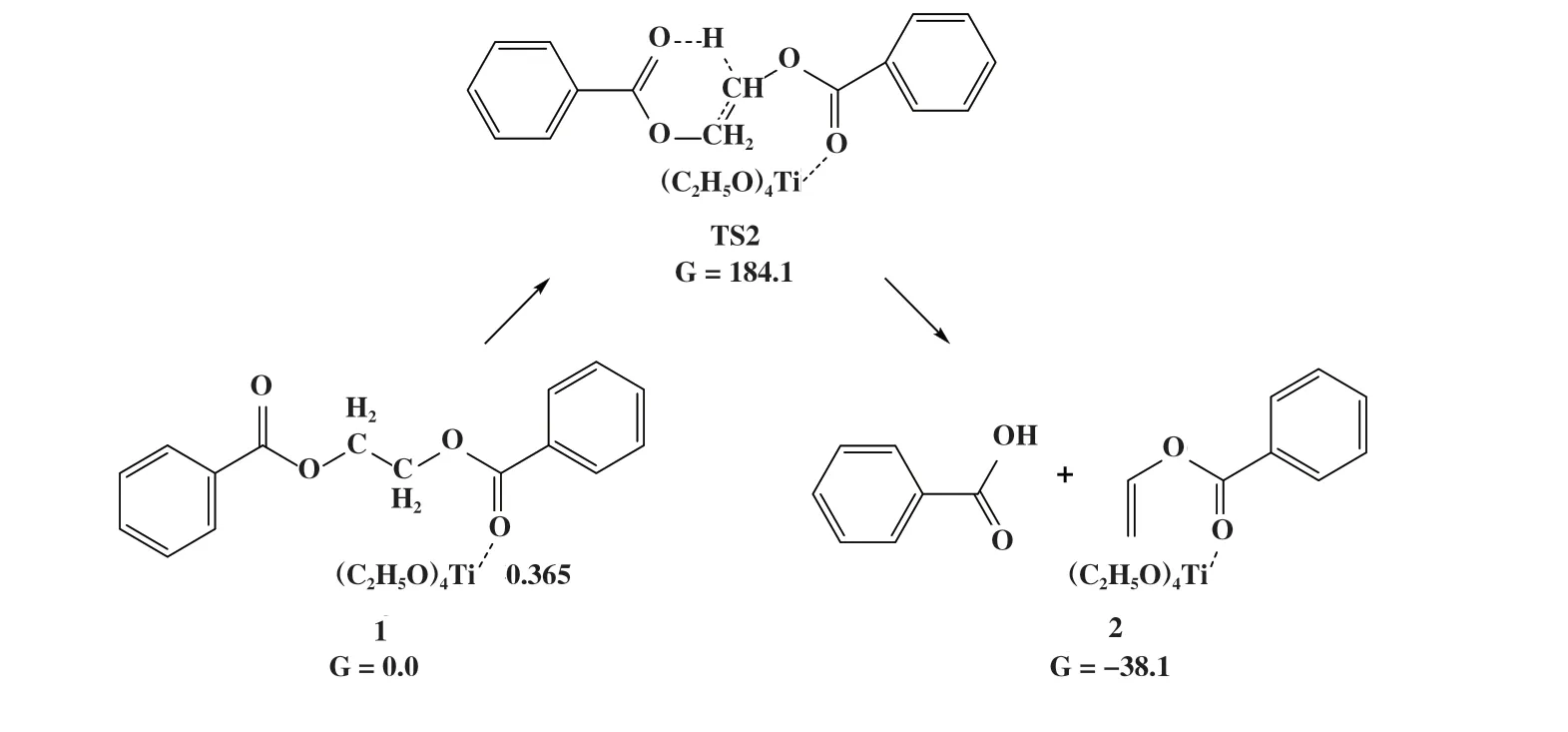
图5 Ti(OEt)4催化EDB热降解的Lewis酸机理Fig.5 Mechanism of Lewis acid-catalyzed EDB thermal degradation reaction for Ti(OEt)4
由于Ti(OEt)4的Lewis酸催化机理无法有效地降低聚酯发生热降解的反应能垒, 而实验报道钛系催化剂对PET的热降解反应具有较明显的促进作用[10],因此我们进一步考察了另一种β消除反应机制,即Ti(OEt)4催化剂中的烷氧基端基氧直接夺取聚酯中乙二醇链段的β-H原子(M2机理), 如图6所示.
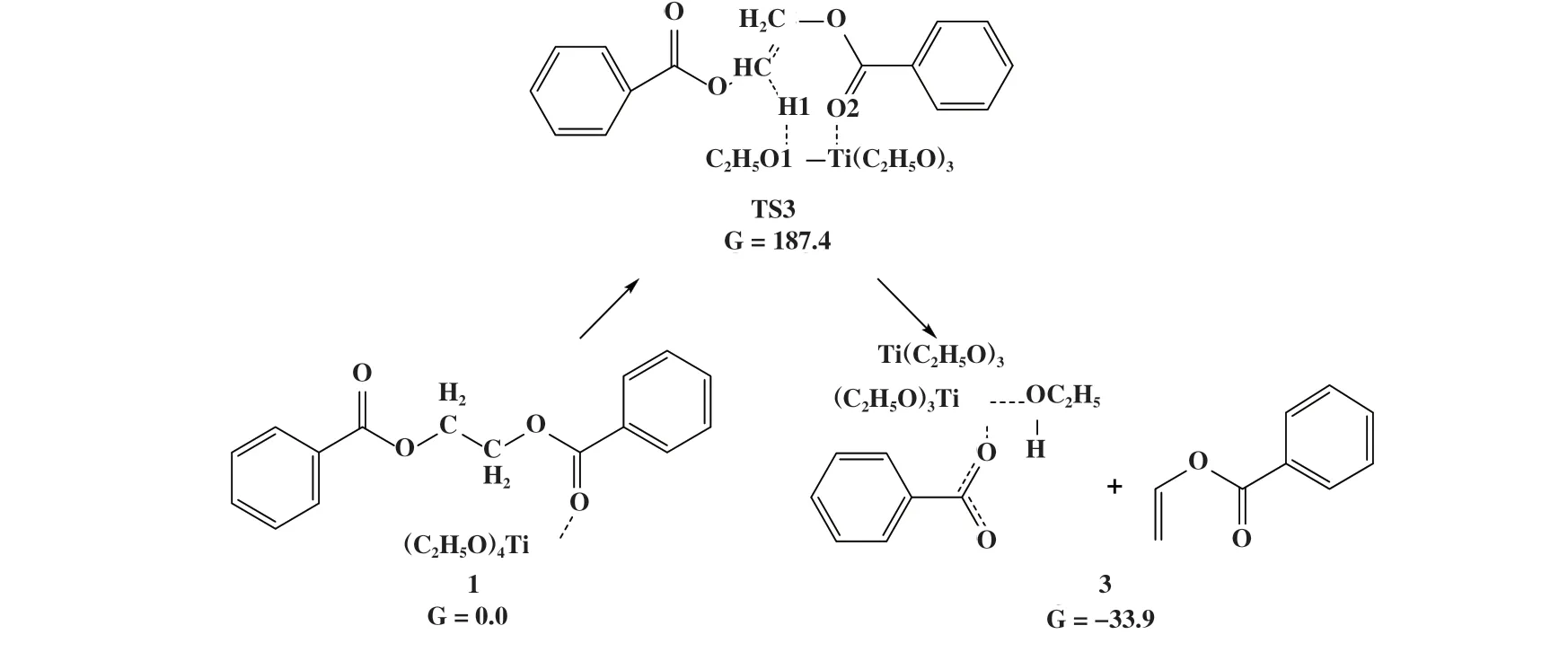
图6 Ti(OEt)4催化EDB热降解烷氧基机理Fig.6 Mechanism of alkoxy-catalyzed EDB thermal degradation reaction for Ti(OEt)4
在M2热降解机理中, 催化剂的烷氧基配体在反应中起到亲核试剂的作用. 基于DFT计算结果, 该产物比反应物能量低33.9 kJ/mol, 表明烷氧基催化聚酯热降解在热力学上有利于反应正向进行. 根据M2反应机理, 烷氧基催化聚酯热降解需要克服187.4 kJ/mol的高能垒, 与无催化剂时的β消除反应活化能(188.3 kJ/mol)相近, 说明在M2 机理中Ti(OEt)4对热降解反应同样未表现出显著的促进作用.
2.4 Ti(OEt)3+催化聚酯热降解反应机理
计算结果表明, 在以Ti(OEt)4为模型催化剂情况下, Lewis酸催化(M1 机理)和烷氧基催化(M2 机理)均无法有效促进聚酯的热降解反应. 通过针对Ti(OEt)4的分子结构分析可知, 由于钛中心和4 个乙氧基配体形成了稳定的八面体结构, 导致反应物EDB不能与钛中心发生有效的相互作用. 由于聚酯热降解的体系中存在一定的酸性物质, H+可以进攻与金属中心配位的一个乙氧基, 从而形成三配位的钛阳离子中心(图4). 为了理解Ti(OEt)3+阳离子中心对聚酯热降解反应的催化性能, 我们同样考察了Lewis酸催化(M1机理)和烷氧基催化(M2机理)两种反应路径. 如图7所示, 反应物4中Ti-O键的键长为0.203 nm. 根据M1机理进行DFT计算的结果表明,产物比反应物能量低38.1 kJ/mol, 且反应需要克服178.2 kJ/mol的能垒完成酯键的断裂. 与无催化剂时的β消除反应能垒(188.3 kJ/mol)相比略有降低, 说明在M1机理中T(iOEt)3+阳离子中心对聚酯的热降解反应存在一定的促进作用.
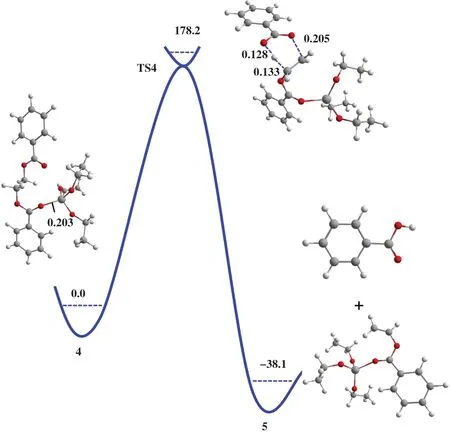
图7 T(iOEt)3+催化EDB热降解的Lewis酸机理Fig.7 Mechanism of Lewis acid-catalyzed EDB thermal degradation reaction for T(iOEt)3+
如图8所示, 根据M2热降解机理计算T(iOEt)3+催化EDB发生分子内β消除反应的能垒为156.1 kJ/mol. 与无催化剂时发生β消除反应的能垒(188.3 kJ/mol)相比有明显下降, 说明在M2机理中T(iOEt)3+阳离子中心对聚酯的热降解反应具有明显的促进作用. 在反应物中, H1-O1的键长为0.233 nm, C1-H1的键长为0.109 nm. 随着反应的进行, 反应物中的C1-H1 键发生断裂, H1 原子逐步转移至O1 原子, H1-O1键长为0.147 nm时体系能量达到最高点,即过渡态结构(图8(b)). 随后, C1-H1键完全断裂,体系能量逐渐降低并形成相应产物.

图8( a)T(iOEt)3+催化EDB热降解烷氧基机理反应过渡态;( b)键长沿内禀反应坐标的变化Fig.8( a) Transition state of alkoxy-catalyzed EDB thermal degradation reaction for T(iOEt)3+;( b) Bond lengths of key atoms along the intrinsic reaction coordinate
2.5 反应机理路径对比分析
如表1所示, 在M1机理和M2机理下, T(iOEt)4催化剂对聚酯热降解反应未表现出明显的促进作用. 这是由于Ti金属中心配位饱和, 导致反应物EDB不能有效地与Ti中心发生相互作用. 而T(iOEt)3+催化剂在M2机理下可以有效促进聚酯进行热降解反应, 反应能垒从无催化剂条件下的188.3下降至156.1 kJ/mol.
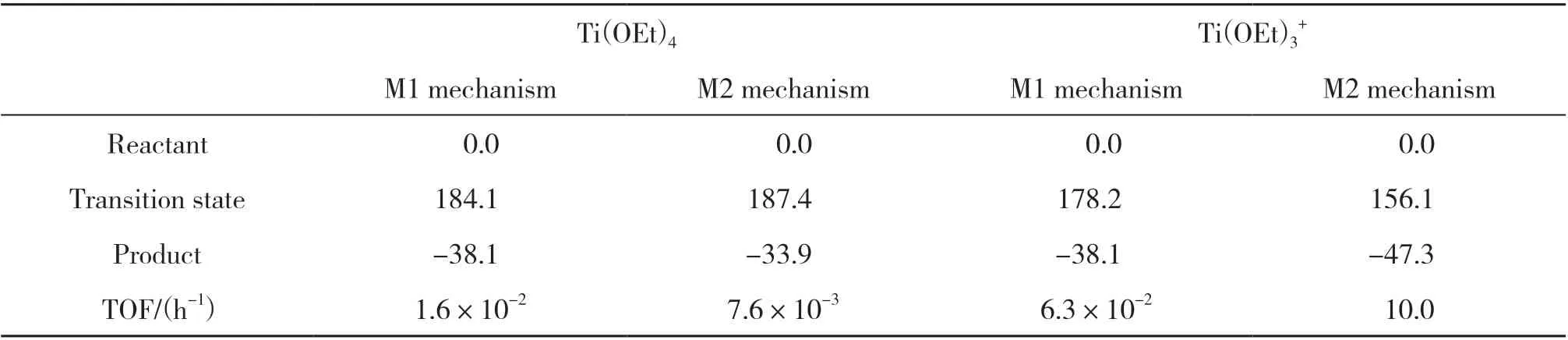
表 1 不同钛系催化剂下β 消除反应及其对应的吉布斯自由能Table 1 β-Elimination reactions of different titanium catalysts and corresponding Gibbs free energies (kJ/mol)
催化反应的转化频率(TOF)是催化剂效率的直观表达. Sebastian等[14]提出, 一个催化反应路径的TOF值通常可由该反应路径的自由能变化进行计算, 表达式如下:

式中, Ta和Ib分别对应反应路径中过渡态和中间体的Gibbs自由能;ΔGr0是反应路径的自由能变,取决于反应物和产物的能量差.
我们根据计算得到的各反应路径能量计算了相应的TOF值, 结果列入表1. 显然, Ti(OEt)3+在M2机理下具有最高的TOF值, 为10.0 h-1. 该结果说明Ti-(OEt)催化剂可能是聚酯热降解反应的催化活性中心, 并且热降解反应遵从M2机理.
为了理解M2机理路径中, Ti(OEt)4催化剂和阳离子Ti(OEt)3+催化剂反应特性的差异, 我们对过渡态TS3和TS5进行了主相互作用轨道(PIO)分析[15],结果如图9所示. PIO分析将两个片段之间的总相互作用分解为成对的轨道相互作用, 并通过PIO的键指数PBI来量化每个轨道相互作用的大小. Ti(OEt)4催化剂与反应物EDB有4对主相互作用轨道, 其中第2对与第3对为金属中心的d轨道作为电子供体向EDB进行电荷输送, 但第4 对的相互作用较弱.在该过渡态TS3 中, Ti(OEt)4催化剂和EDB之间的总PBI值为1.10. 阳离子Ti(OEt)与反应物EDB同样具有4对主相互作用轨道, 其中3对金属中心的d轨道皆为强作用相互轨道. 由于具备更强的轨道相互作用, 催化剂和EDB之间的总PBI值为1.27, 高于过渡态TS3. 因此, 过渡态TS5的化学稳定性更高,说明在M2热降解机理下, 阳离子Ti(OEt)3+催化剂能更有效地催化EDB发生分子内β消除反应.
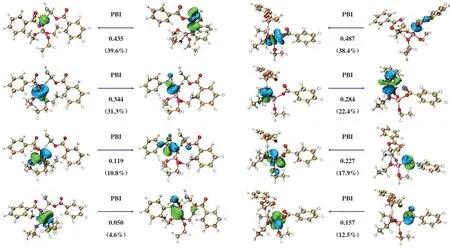
图9 过渡态TS3和TS5的主相互作用轨道分析Fig.9 Principal interacting orbital (PIO) analysis of TS3 and TS5
3 结论
我们主要考察了两种钛系催化剂催化聚酯热降解的反应机理, 其主要结论如下:( 1)Lewis 酸催化热降解反应机理在钛系催化剂下的解聚反应能垒与无催化剂反应相近, 未表现出明显促进作用;( 2)阳离子T(iOEt)3+催化剂可能是聚酯热降解反应的催化活性中心, 并且热降解反应遵从M2机理;( 3)T(iOEt)3+和反应物 EDB 之间具有更高的 PBI 值, 过渡态 TS5的稳定性更高, 有利于EDB发生分子内β消除反应.
钛系聚酯催化剂在工业生产中通常需要加入含磷化合物作为稳定剂, 用来改善钛系催化剂聚酯产品的品质. 根据我们的研究结果, 含磷稳定剂可能起到稳定T(iOEt)3+阳离子中心的作用, 抑制了钛系催化剂催化聚酯过程中催化热降解副反应的发生.我们的研究结果对开发新型钛系催化剂具有一定的理论指导意义.
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
北京航空航天大学学报(2022年5期)2022-06-06
华东理工大学学报(自然科学版)(2022年2期)2022-04-29
浙江大学学报(理学版)(2021年6期)2021-12-02
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
