人类卵母细胞减数分裂的生理和病理机制
2023-12-20 09:12周舟桑庆王磊
遗传 2023年12期
周舟,桑庆,王磊
特邀综述
人类卵母细胞减数分裂的生理和病理机制
周舟1,2,桑庆1,王磊1
1. 复旦大学生物医学研究院,上海 200032 2. 上海市生物医药技术研究院,上海 200237
正常的卵子发生是人类成功繁育后代的关键步骤。女性胚胎发育时期,原始生殖细胞从有丝分裂转变为减数分裂,经过同源染色体配对和重组后,减数分裂被阻滞在减数第一次分裂前期的双线期。卵泡内卵母细胞的减数分裂阻滞的维持主要归因于胞质中高浓度的环磷酸腺苷。在月经周期中,卵泡刺激素和黄体生成素促进某些卵母细胞恢复减数分裂,完成排卵过程。卵母细胞减数分裂过程中发生任何缺陷都可能影响卵子发生,进而影响受精和胚胎发育过程。辅助生殖、高通量测序和分子生物学技术的快速发展,为人类认识减数分裂背后的精确分子机制以及卵母细胞成熟缺陷疾病的发病机制与诊疗提供新的思路和手段。本文主要介绍了近年来发现的调控卵子发生的生理和病理机制,涉及同源重组、减数分裂阻滞与恢复、母源mRNA降解、翻译后调节、透明带组装等过程,旨在增进相关领域研究人员对卵母细胞减数分裂的了解,并为进一步机制研究和疾病治疗提供理论基础。
卵子发生;卵母细胞;减数分裂;突变
成熟的卵母细胞,被称为卵子,是终末成熟状态的女性生殖细胞,由卵原细胞通过减数分裂而产生[1]。卵子发生,即卵母细胞的生长和分化过程,不仅对受精和胚胎发育至关重要,而且对胎儿后期的发育和生长也有长期的影响。这一过程的分子调控也是生殖医学及发育生物学最重要的科学问题之一。
卵母细胞减数分裂过程至少在3个方面与精子不同:女性生殖细胞在胚胎发育过程中进入并经历减数第一次分裂进程,并且在出生前停滞在减数第一次分裂前期的双线期阶段;在青春期后的每个月经周期中,一些在完全生长的卵泡内停滞的卵母细胞将恢复减数分裂;卵母细胞的细胞分裂为不对称胞质分裂。在激素的作用下,初级卵母细胞恢复减数分裂,发生染色质浓缩、细胞核膜溶解,进入减数第一次分裂中期;随后,卵母细胞排出含有一小部分细胞质和1组染色体的第一极体,完成减数第一次分裂;不久之后,卵母细胞开始减数第二次分裂,停滞于细胞分裂中期;受精后,卵母细胞才完成减数分裂过程[1]。卵母细胞的减数分裂成熟包括一系列复杂的细胞核和细胞质事件,最终获得卵母细胞以及早期胚胎发育所需的全部能力。
近年来,得益于辅助生殖技术的发展和分子生物学技术的进步,科学家们利用人类卵母细胞样本和转基因小鼠模型发现精确调控减数分裂成熟的多种信号通路[2,3]。任何涉及这些关键反应或信号途径的干扰都会造成低质量卵子,进而导致受精失败或早期胚胎停滞等异常生殖表型[4]。本文讨论了从卵原细胞发育为卵子的重要生理调节机制以及导致人类卵母细胞成熟缺陷的病理因素,为进一步研究卵母细胞减数分裂奠定基础。
1 卵母细胞减数分裂过程
卵子发生在女性胚胎发育早期就已经开始[5],大约在胚胎发育第7周,卵原细胞首先出现在性腺脊上。卵原细胞与外面包裹的单层扁平颗粒细胞共同组成原始卵泡。直至妊娠第20周,原始生殖细胞经历快速的有丝分裂,产生600~700万个卵原细胞,这些卵原细胞陆续分裂分化为初级卵母细胞,随后进入减数分裂过程。减数分裂是卵子发生的关键环节,分为前期、中期、后期、末期4个阶段,减数第一次分裂前期根据染色体形态分为细线期、偶线期、粗线期、双线期和终变期。同源染色体在偶线期联会配对并在粗线期重组。卵母细胞的数量通过卵泡闭锁持续减少,直到出生时新生儿只有100~200万个初级卵母细胞。这些卵母细胞停滞于减数第一次分裂前期的双线期,因细胞核膨大而被称为生发泡(germinal vesicle,GV)卵母细胞,其后进入漫长的静息阶段,直到青春期来临才恢复减数分裂[1]。
女性青春期后,卵巢由于卵泡闭锁只剩下30~50万个卵泡,部分原始卵泡通过磷脂酰肌醇3激酶/蛋白激酶B(phosphoinositide 3-kinase/protein kinase B,PI3K/Akt)信号通路的激活和抗缪勒管激素(anti- Müllerian hormone,AMH)的抑制而脱离静息状态,开始生长发育并依次形成初级和次级卵泡[6,7]。在此期间,颗粒细胞快速分裂,形成多层颗粒细胞层,为卵母细胞提供多种生长必需的物质;初级卵母细胞的生发泡增大,进入转录活跃的网状期,逐渐积累mRNA和蛋白等物质,导致卵母细胞直径增大、并且卵外周形成一层被称为透明带的细胞外基质[8]。在垂体分泌的卵泡刺激素(follicle-stimulating hormone,FSH)和黄体生成素(luteinizing hormone,LH)的作用下,次级卵泡发育为窦状卵泡,最终形成一个最大的优势卵泡并排卵[6,9]。卵泡生长基本完成到排卵之间仅有数天时间。首先,初级卵母细胞恢复减数分裂,染色质浓缩、细胞核膜溶解,该过程通常被称为生发泡破裂(germinal vesicle breakdown,GVBD)。随后,初级卵母细胞的纺锤体组装,使染色体整齐排列在细胞中央,进入减数第一次分裂中期(metaphase I,MI),然后经过不均等分裂产生次级卵母细胞和第一极体。次级卵母细胞几乎包含全部的初级卵母细胞细胞质,极体则是无分裂和受精能力的单倍体小细胞,通常依附于卵母细胞的动物极(图1)。最后,次级卵母细胞的纺锤体在极短的时间内迅速解聚并再次聚合,引导剩余染色体发生赤道板集合,进入减数第二次分裂中期(metaphase II,MII)并再次静止,这一时期的MII卵母细胞通常被称为卵子(图1)[8]。卵子透明带之外围绕着若干层卵丘颗粒细胞,最靠近透明带的一层被称为放射冠,具有保护卵母细胞和提供营养物质的作用。女性排卵时,卵泡破裂,卵子连同周围的透明带和放射冠组成卵丘-卵母细胞复合体(cumulus-oocyte complex,COC),随卵泡液一起从卵巢排出,被输卵管伞端所拾取,由于输卵管上皮细胞纤毛的摆动和肌层的收缩,进入输卵管壶腹部,等待精子受精[10]。卵子在受精之后发生一系列生理生化变化,包括钙离子震荡、减数分裂恢复、皮质反应、雌雄原核形成等,通过不均等分裂排出第二极体(图1),完成减数第二次分裂;若未受精,该次级卵母细胞将在排卵后12~24 h内退化[11]。因此,哺乳动物中不存在真正意义上单倍体的卵子。女性一生中只有400~500个卵泡能够成为优势卵泡,从而完成排卵过程[12]。

图1 人类卵母细胞减数分裂过程
GV代表生发泡,GVBD代表生发泡破裂,MI代表减数第一次分裂中期,MII代表减数第二次分裂中期,2PN代表两原核。
2 卵子发生的调控机制
卵子发生包括同源重组、减数第一次分裂阻滞和恢复、减数第一次分裂向减数第二次分裂转变、减数第二次分裂中期阻滞等关键过程。丝裂原激活蛋白激酶、成熟促进因子、减数分裂抑制因子、Securin、分离酶等多种蛋白可通过泛素-蛋白酶体通路、磷脂酰肌醇通路、环磷酸腺苷通路等多个信号通路调控卵母细胞的成熟。这些通路中关键蛋白的研究建立了系统的卵子发生调控机制,为减数分裂缺陷的病理机制研究提供了理论基础。
2.1 程序化的DNA双链断裂
同源重组是减数分裂所特有的核心步骤,保证了后代遗传物质的重新分配、同源染色体的精确分离及遗传多样性[13]。每对同源染色体由两个通过黏附蛋白连接的姐妹染色单体组成,程序化的DNA双链断裂(DNA double-strand breaks,DSBs)诱导姐妹染色单体之间产生交换重组、交叉重组或非交叉重组,交叉及非交叉重组都会导致同源染色体之间片段发生交换[14]。DSB位点并非沿染色体随机分布,减数分裂特异的甲基转移酶PRDM9 (PR/SET domain 9)通过锌指结构域结合DNA并进行组蛋白第3亚基4号赖氨酸的三甲基化(H3K4me3)和组蛋白第3亚基36号赖氨酸的三甲基化(H3K36me3)修饰,从而确定DSB热点区域[15]。HORMAD1 (HORMA domain containing 1)蛋白优先结合未联会的染色体轴,通过招募IHO1 (interactor of HORMAD1 1)实现DSBs,促进蛋白IHO1、REC114 (REC114 meiotic recombination protein)和MEI4 (meiotic double- stranded break formation protein 4)在染色轴上组装为三聚体复合物,将拓扑异构酶SPO11 (SPO11 initiator of meiotic double strand breaks)与染色体轴联系起来[16]。在PRDM9甲基化作用和IHO1- REC114-MEI4的帮助下,SPO11被募集到染色体轴处共价结合DNA并发挥作用。此后核酸内切酶MRE11 (MRE11 double strand break repair nuclease A)将SPO11-寡核苷酸复合物从染色质上切除并导致DSBs位点暴露短的单链DNA尾巴[17]。这些单链尾巴通过核酸酶进一步延伸,最终结合重组酶DMC1 (DNA meiotic recombinase 1)和RAD51 (RAD51 recombinase)完成同源重组[18]。
2.2 卵母细胞GV阻滞维持和减数分裂恢复
成熟促进因子(maturation promoting factor,MPF)在GVBD和随后的卵母细胞成熟过程中发挥着关键作用。复合物MPF由催化亚基细胞周期蛋白依赖性激酶CDK1 (cyclin dependent kinase 1,别名CDC2)和调节亚基细胞周期蛋白Cyclin B组成,磷酸化其靶蛋白的特定丝氨酸和苏氨酸残基,以发挥激酶活性。在卵母细胞发育早期,卵泡内的膜细胞、壁颗粒细胞和卵丘颗粒细胞产生环磷酸腺苷(cyclic adenosine monophosphate,cAMP)并持续转移到卵母细胞中,从而保持卵母细胞内高水平的cAMP[19]。高水平的cAMP持续激活蛋白激酶A (protein kinase A,PKA),PKA磷酸化并激活蛋白激酶WEE2 (WEE2 oocyte meiosis inhibiting kinase),激活的WEE2蛋白能够抑制磷酸酶CDC25 (cell division cycle 25),而CDC25是CDK1的激活剂;同时,蛋白激酶WEE2将CDK1的Thr14和Tyr15磷酸化而抑制其活性(图2A)。此时,身为泛素连接酶的后期促进复合体(anaphase promoting complex/cyclosome,APC/C)持续降解Cyclin B蛋白(图2A)。以上数个信号通路使MPF保持在失活状态,而低活性的MPF有利于维持磷酸酶1 (phosphatase 1,PP1)对减数分裂蛋白的去磷酸化作用,进而使卵母细胞停滞在GV时期[19]。
在优势卵泡的颗粒细胞内,LH结合相应受 体LHCGR (luteinizing hormone/choriogonadotropin receptor),产生cAMP并激活PKA,促进表皮生长因子(epidermal growth factor,EGF)相关多肽的释放[20]。这些因子结合EGF受体,从而激活RAS和细胞外信号调节激酶(extracellular signal-related kinase 1/2,ERK1/2)信号通路[20]。ERK1/2一方面减弱了环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)介导的减数分裂抑制信号;一方面磷酸化缝隙连接蛋白CX37 (Connexin 37),阻碍cAMP和cGMP在颗粒细胞和卵母细胞间的传递[21,22]。当卵母细胞中的cAMP水平降低,WEE2被从核中运出,激活的CDC25将CDK1去磷酸化;同时,FBXO5表达升高并抑制APC/C的活性以积累Cyclin B;导致MPF恢复活性,调节下游多种信号通路,进而促使卵母细胞GVBD (图2B)[19]。
ERK1/2在GVBD后促进纺锤体的组装,从而调控卵母细胞减数分裂周期进程。ERK1/2能够磷酸化细胞质中的RNA结合蛋白CPEB1 (cytoplasmic polyadenylation element binding protein 1),激活的CPEB1蛋白结合到mRNA的3′UTR区域的CPE (cytoplasmic polyadenylation element)位点,从而延长纺锤体组装相关转录本的poly(A)尾并启动其翻译[23]。当卵母细胞完成减数第一次分裂后,立即开始减数第二次分裂,并停滞在MII阶段。
2.3 卵母细胞MII阻滞和减数分裂恢复
MII卵母细胞通过细胞静止因子(cytostatic factor,CSF)信号通路维持MPF的活性,而停滞在MII阶段(图3)。CSF由多个分子或蛋白组成,CSF的重要组分FBXO43 (F-box protein 43)通过结合APC/C效应器CDC20 (cell division cycle 20)而抑制APC/C的活性,有利于Cyclin B和分离酶抑制剂Securin的积累[24]。MOS (MOS proto-oncogene, serine/threonine kinase)诱导的丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)激活能够稳定FBXO43蛋白[25];同时,MOS/MEK/MAPK/p90RSK级联通路可以维持CSF的功能,从而抑制APC/C的活性,协助卵母细胞MII阻滞[26]。
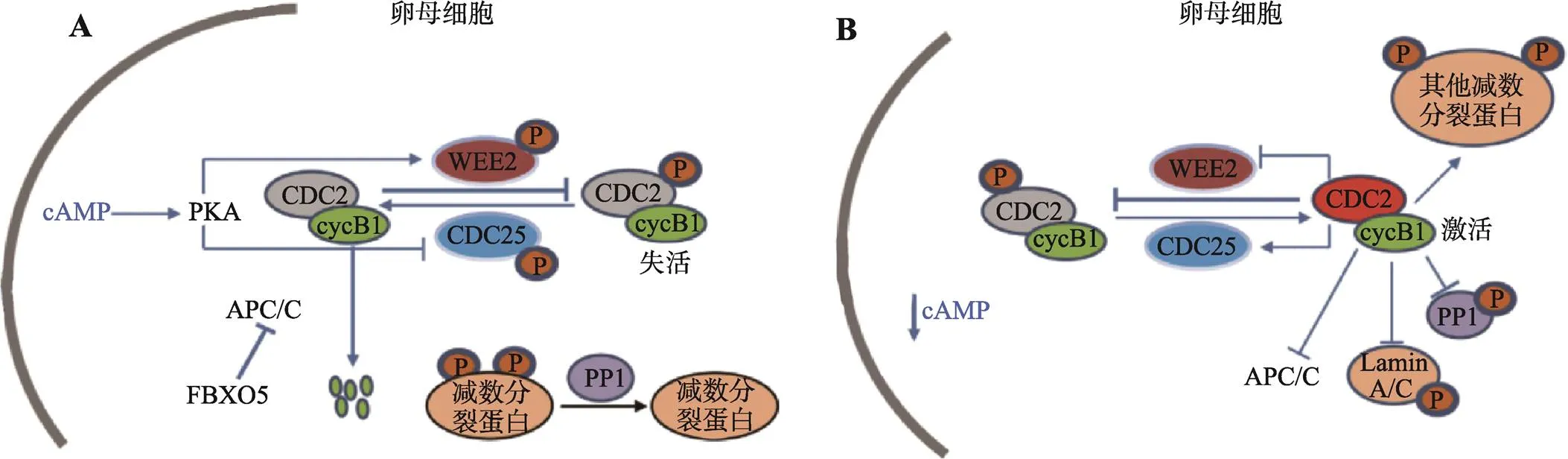
图2 减数第一次分裂阻滞和恢复的调控机制
A:减数第一次分裂前期阻滞的调控机制;B:减数第一次分裂恢复的调控机制。
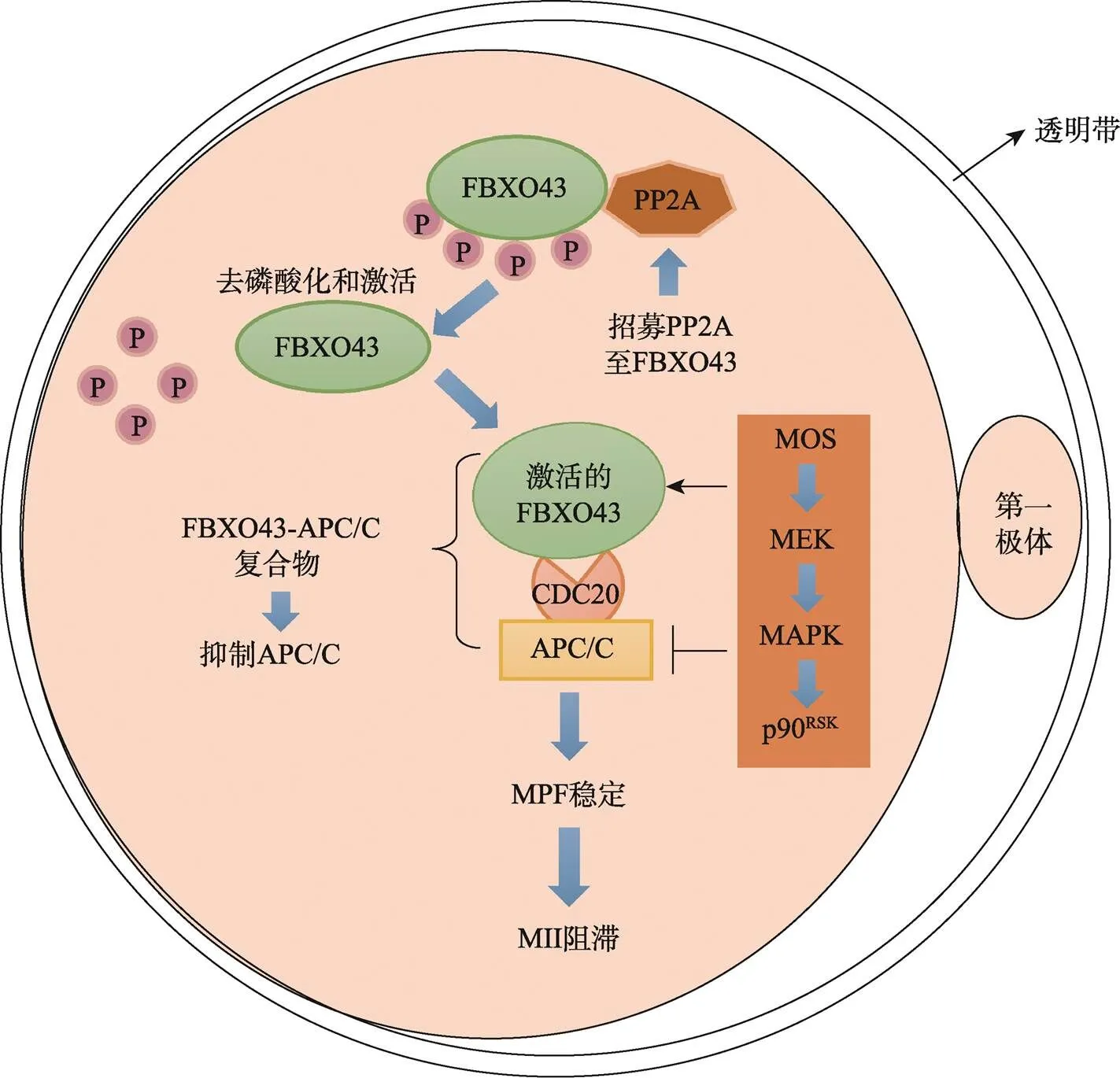
图3 减数第二次分裂阻滞的调控机制
精子进入卵子后,释放一系列信号,引发钙离子震荡,钙离子作为第二信使与钙调蛋白结合,激活钙离子/钙调蛋白依赖性蛋白激酶II (calcium/ calmodulin dependent protein kinase II,CAMKII),CAMKII活化WEE2,WEE2磷酸化CDK1,使MPF活性降低;同时,CAMKII导致FBXO43磷酸化失活和降解,激活APC/C,无法维持MPF的稳定,使Cyclin B被降解;最终促使卵子重新进入减数分裂,姐妹染色单体分离并排出第二极体[27]。
3 卵母细胞减数分裂缺陷
人类卵母细胞的减数分裂成熟过程需要经过一系列的分子和形态变化,一般包括两部分:核成熟和质成熟[28]。核成熟主要发生在LH水平剧增之后,GV卵母细胞重新进入减数第一次分裂,染色体在纺锤体的作用下有序分离并排出卵母细胞[8]。而质成熟则提供核成熟过程所涉及到的一些细胞关键组分并调控相应分子的合成,为卵子受精和胚胎发育提供物质准备[28]。卵子发生过程中初级卵母细胞始终无法排出第一极体,即核成熟缺陷,被称为卵母细胞成熟障碍,根据卵母细胞发育停滞的不同时期,分为GV阻滞和MI阻滞。目前临床上仅靠形态学判断精子、卵子以及胚胎质量,然而形态上的正常并不意味着精子、卵子和胚胎在关键性的分子水平上是正常的,即形态正常的精子和卵子可能由于质成熟缺陷而弱化或失去发育潜能,因此无法顺利完成受精及胚胎发育过程。只有拥有完整发育潜能的卵子才能正常受精,而在卵母细胞减数分裂成熟过程中出现任何缺陷和错误都将可能导致成熟障碍,或者影响其后的受精和胚胎发育过程,最终导致受精失败和早期胚胎停滞[28]。受精失败根据临床特征被划分为精卵结合异常、无法形成原核和原核数目异常。早期胚胎停滞表型指移植前早期胚胎停止分裂而无法形成囊胚或者囊胚无法着床。其中,合子分裂失败是早期胚胎停滞的一种独特表型,表现为卵子受精后合子不卵裂。
在临床实践中,许多女性不孕患者的卵巢储备、月经周期、子宫及双侧附件正常,基础性激素水平和其他不孕症相关的检查未见明显异常;然而,她们因遗传学因素而患有卵母细胞成熟缺陷,具体表现为卵子死亡、透明带异常、卵母细胞成熟障碍、受精失败和早期胚胎停滞等,经过多次辅助生殖治疗仍无法成功妊娠,饱尝多个辅助生殖周期失败的经济和精神压力、以及反复促排卵过程对身体的伤害[4]。
4 卵母细胞减数分裂异常的病理机制
近年来,一些涉及调控卵母细胞减数分裂的基因(包括、、、、、、、、、、、、、、、、、、、、、、和)被报道与人类一系列卵母细胞成熟缺陷表型相关,包括卵母细胞成熟障碍、受精失败、早期胚胎停滞和胚胎着床失败(表1)。
4.1 突变破坏同源重组
(thyroid hormone receptor interactor 13)广泛表达于包括生殖细胞在内的各种组织中,在有丝分裂和减数分裂中均发挥重要作用。在减数分裂过程中,主要在减数第一次分裂前期发挥功能,参与同源染色体联会和重组。TRIP13是检验点蛋白HORMAD1和HORMAD2(HORMA domain containing 2)的负调控因子,通过调控HORMAD2从突触染色体轴上移除而打开检验点,使同源染色体得以成功分离。错义突变降低了TRIP13蛋白水平,并导致其下游分子HORMAD2的累积,阻碍了同源染色体的分离,最终造成卵母细胞发育缺陷。携带纯合或复杂合错义突变的患者均表现为卵母细胞MI阻滞或合子分裂失败[29]。
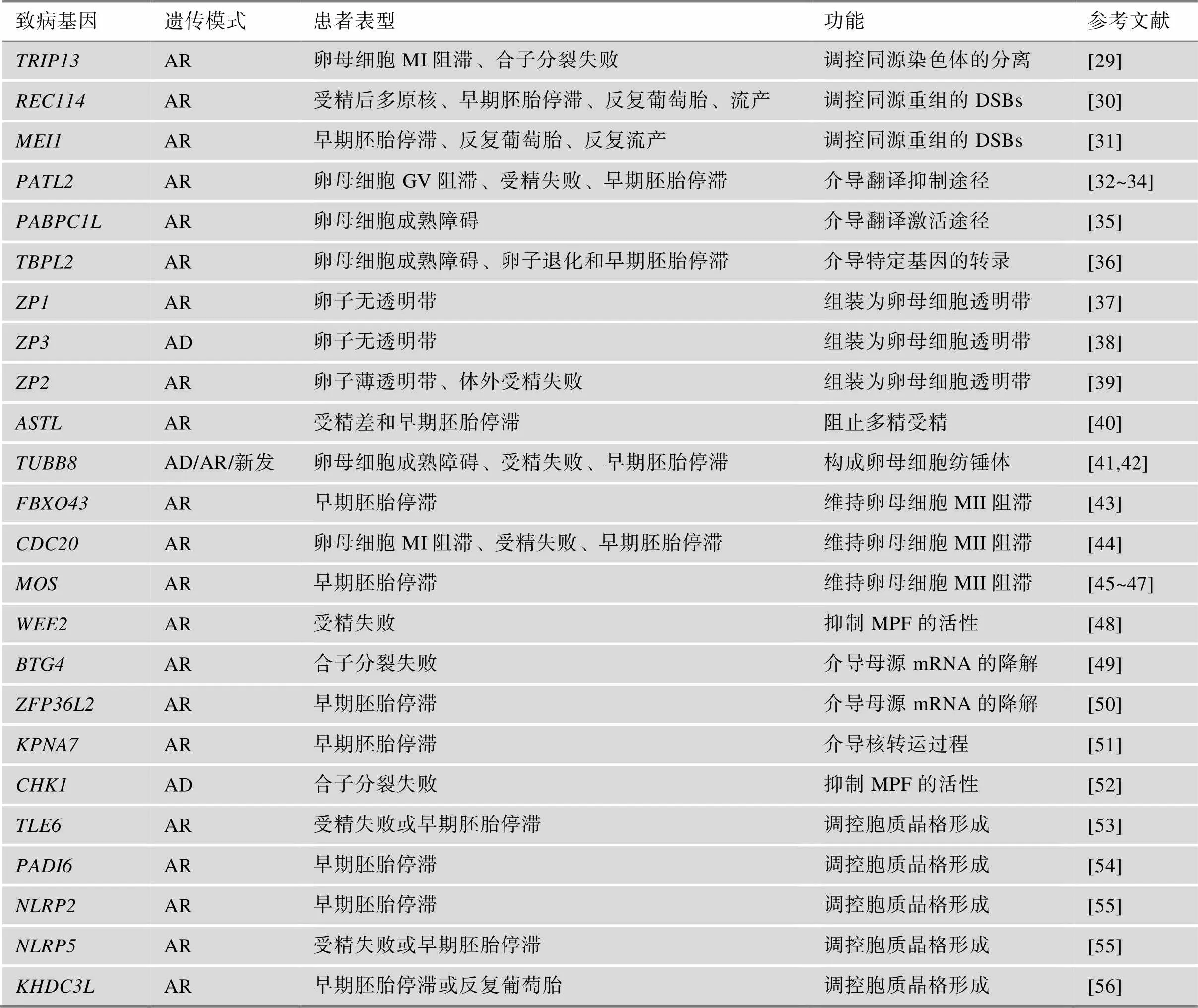
表1 卵母细胞减数分裂缺陷的致病基因
AD:常染色体显性遗传;AR:常染色体隐性遗传;GV:生发泡;MI:减数第一次分裂中期;MII:减数第二次分裂中期;DSBs:DNA双链断裂;MPF:成熟促进因子。
突变导致自身蛋白水平降低,并且失去了维持MEI4稳定性的能力,从而导致REC114- MEI4-IHO1复合物功能丧失。另外,REC114参与促进SPO11与染色质的结合,MEI4的正常染色体定位受REC114和MER2影响。因此,REC114蛋白量减少可能导致SPO11和MEI4无法正确定位,造成染色体配对过程中DSBs发生异常。异常DSBs所诱导的同源染色体重组可能会进一步造成染色体排列混乱或分离异常,导致卵母细胞发育缺陷,具体表现为受精多原核、早期胚胎停滞、反复葡萄胎和流产,表型差异可能取决于不同突变带来的不同影响[30]。
MEI1 (meiotic double-stranded break formation protein 1)蛋白是正常减数分裂染色体联会所必需的,并且在减数分裂DSBs的形成中发挥关键作用。隐性突变破坏了MEI1蛋白的功能,进而导致早期胚胎停滞、反复葡萄胎或反复流产[31]。
4.2 突变影响母源mRNA翻译
随着卵泡发育,生发泡卵母细胞逐渐积累了大量处于翻译抑制状态的母源性mRNA。在卵子发生过程中,卵胞质储存的母源mRNA开始有序的分批次被翻译加工,主要参与调控纺锤体组装、MII阻滞的维持以及部分母源mRNA降解等过程,对于卵子发生和早期胚胎发育起着关键性的作用[57,58]。在人类卵子中,(PAT1 homolog 2)突变导致相应蛋白快速降解或破坏,卵子内PATL2蛋白减少可能扰乱了正常的翻译抑制途径并激活下游的一些转录本提前进入蛋白合成过程[32]。因此,突变除了导致本身蛋白功能丧失之外,还有一个可能的致病机制是破坏了其他mRNA结合蛋白的总量,从而导致患者的卵母细胞GV阻滞、受精失败及早期胚胎停滞表型[33,34]。另外,患者表型的多样性取决于突变对蛋白的破坏程度,破坏性越大,则卵子发育阻滞在更早期的阶段。
PABPC1L (poly(A) binding protein cytoplasmic 1 like)是人类卵母细胞和合子基因组激活前早期胚胎中的一种主要poly(A)结合蛋白,在母源mRNA的翻译激活中起着关键作用。突变通过影响PABPC1L与母源mRNA的结合,导致蛋白截短、蛋白水平降低、定位改变以及母源mRNA翻译激活减少,从而影响卵母细胞的胞质成熟,导致卵母细胞成熟障碍[35]。
TBPL2 (TATA-box binding protein like 2)是卵母细胞特异性的通用转录因子,可以结合活性基因启动子的TATA盒并介导RNA聚合酶II的转录起始。纯合剪接突变破坏了mRNA的结构,导致密码子提前终止,因此降低了许多对卵母细胞成熟和受精很重要的卵母细胞特异性基因的表达,从而导致卵母细胞成熟障碍(包括GV和MI阻滞)、卵子退化和早期胚胎停滞[36]。
4.3 突变破坏透明带结构
透明带能够协助卵母细胞生长和卵泡发育、辅助精卵相互识别和特异性结合、限制异种受精和多精受精、保护早期胚胎[59]。在人类卵泡发育过程中,生长中的初级卵母细胞表达糖蛋白ZP1 (zona pellucida glycoprotein 1)、ZP2 (zona pellucida glycoprotein 2)、ZP3 (zona pellucida glycoprotein 3)和ZP4(zona pellucida glycoprotein 4)并分泌到胞外。这些糖蛋白相互交联,从而形成透明带。隐性突变可能会影响ZP1蛋白在体内的表达或分泌,从而导致透明带纤维无法相互连接,阻止透明带的形成[37]。基因杂合突变通过显性负效应影响野生型透明带蛋白间的相互作用,从而阻碍透明带的有序组装[38]。当透明带缺失时,卵丘颗粒细胞将完全脱离卵子,导致卵子退化消失。基因纯合突变破坏了ZP2蛋白的表达和分泌,突变型ZP2蛋白无法分泌到卵子表面,因而患者卵子周围只能形成一层薄而有缺陷的仅由另外3种透明带蛋白组成的透明带[39]。精子与患者卵子薄透明带的结合缺陷是体外受精失败的原因,最终严重影响女性的生育能力。
4.4 突变影响透明带切割
精卵融合时,卵胞质的皮质颗粒中的水解酶通过胞吐作用释放到卵周隙,其中ASTL (astacin like metalloendopeptidase)蛋白切割透明带表面结合精子的ZP2蛋白,导致其他精子不能识别与结合卵子,继而诱导透明带硬化,阻止其他精子穿过透明带。突变破坏了ASTL的结构,ASTL缺失的卵子仍然能够在一定程度上阻止多精受精,得到质量较好的8细胞胚胎。但ASTL缺失影响了卵母细胞的胞质成熟,造成患者卵子受精差和早期胚胎停滞的表型,最终导致女性不孕[40]。
4.5 突变破坏纺锤体结构
TUBB8 (tubulin beta 8 class VIII)是灵长类特异性β微管蛋白亚型,特异性表达于卵子和早期胚胎中,是构成卵母细胞纺锤体β微管蛋白的最主要的形式。有丝分裂和减数分裂纺锤体都由微管组成,这些微管是由α微管蛋白和β微管蛋白组装而成的动态聚合物[42]。突变遵循显性或隐性遗传模式,新发和遗传的错义杂合突变通过显性负效应影响依赖伴侣蛋白的折叠、改变微管动力学、使纺锤体形态结构紊乱;而纯合缺失和移码突变通过单倍剂量不足效应破坏微管功能,影响减数分裂纺锤体组装;不同突变导致患者卵子和胚胎发育表型多样化,包括卵母细胞成熟障碍、受精失败、早期胚胎停滞及反复移植失败等,最终造成女性原发性不孕[41]。
4.6 突变影响MII阻滞的维持
FBXO43在人的卵母细胞、睾丸和肝脏组织中高表达,作为APC/C的抑制分子在卵母细胞减数第二次分裂阻滞和早期胚胎发育中发挥重要功能。突变破坏了FBXO43蛋白的正常表达模式,同时降低了FBXO43稳定下游MPF组分Cyclin B的能力,造成患者卵母细胞减数分裂异常,进而影响受精后早期胚胎发育,最终导致女性不孕[43]。
CDC20作为APC/C的效应器在双线期卵母细胞中表达较低,而在阻滞的MII卵子中表达升高。FBXO43结合CDC20后能够抑制APC/C的活性,从而维持MPF的稳定性。携带隐性突变的患者表现出卵母细胞MI阻滞、受精失败或早期胚胎停滞的表型,这取决于不同突变对CDC20蛋白的损伤程度[44]。
基因编码丝氨酸/苏氨酸蛋白激酶,作为细胞生长抑制因子,通过磷酸化丝裂原活化蛋白激酶(mitogen-activated protein kinase,MEK)而激活细胞外调节蛋白激酶(extracellular regulated protein kinase,ERK)通路,从而维持卵母细胞MII阻滞[45]。MOS-ERK信号通路介导的MII阻滞依赖于FBXO43介导的APC/CCDC20抑制以防止Cyclin B降解[45]。mRNA在GV卵母细胞中几乎未被翻译,但在卵母细胞成熟过程中快速翻译并在受精后迅速降解。隐性突变影响蛋白表达和ERK1/2激活。卵母细胞中MOS-ERK信号通路的失活导致F-肌动蛋白组装异常、母源效应基因降解缺陷和线粒体功能障碍,导致早期胚胎停滞[45~47]。
WEE2是一种卵母细胞特异性蛋白激酶,通过调控MPF的活性而调控细胞周期。在人类卵子中,突变破坏了蛋白结构,进而影响蛋白的稳定性和功能,导致突变型WEE2蛋白自身磷酸化水平、磷酸化下游CDK1蛋白Tyr15的能力以及促进卵子生成原核的能力均有显著下降,造成卵子受精后无法形成雌雄原核,即受精失败表型[48]。
4.7 突变影响母源合子转换
母源合子转换是卵母细胞向早期胚胎的基因表达模式转换。其中,卵胞质储存的母源mRNA的适时降解是胚胎自身基因组转录激活的重要前提,对于卵子发生和早期胚胎发育至关重要。脱腺苷酸化酶CCR4-NOT复合物通过脱腺苷酸化缩短母源mRNA的poly(A)尾而开始mRNA降解过程。BTG4 (BTG anti-proliferation factor 4)是一种CCR4-NOT复合物的衔接蛋白,能够结合母源mRNA的5′端m7G帽和3′端poly(A)尾巴上的结合蛋白EIF4E、PABPN1和PABPC1L,进而招募催化亚基CNOT7 (CCR4-NOT transcription complex subunit 7)或CNOT8 (CCR4-NOT transcription complex subunit 8),触发CCR4-NOT复合物介导的母源mRNA降解途径[60]。已报道的基因纯合突变均破坏BTG4蛋白的正常功能,包括蛋白缺失、截短和影响BTG4与CNOT7之间的互作,从而影响母源mRNA降解和母源合子转换,造成患者合子中母源mRNA非正常累积,最终导致合子分裂失败[49]。
在卵母细胞成熟过程中,RNA结合蛋白ZFP36L2 (ZFP36 ring finger protein like 2)通过结合CCR4- NOT复合物的催化亚基CNOT6L而招募该复合物,从而介导具有腺嘌呤/尿嘧啶富集元件(AU-rich element,ARE)的mRNA的降解[61]。人类复杂合突变被报道破坏合子的母源mRNA的降解,从而影响早期胚胎发育,造成早期胚胎停滞[50]。
(karyopherin subunit alpha 7)编码核转运蛋白α7蛋白,KPNA7是人类卵母细胞和早期胚胎中表达最高的核转运蛋白α家族成员,介导核蛋白从细胞质到细胞核的转移。突变降低了KPNA7蛋白水平,减弱了KPNA7与底物RSL1D1 (ribosomal L1 domain containing 1)结合的能力,并影响了KPNA7的核转运活性,从而阻碍某些底物的核输入,而这些底物对于胚胎自身基因组转录激活至关重要[51]。
4.8 突变破坏细胞周期进程
(checkpoint kinase 1)编码一种丝氨酸/苏氨酸蛋白激酶,主要定位于细胞核中的染色质上。CHK1通过N-末端激酶结构域和C-末端调节结构域之间的相互作用而保持闭合构象;而保守基序CM1或CM2中的突变可以破坏CHK1的闭合构象,暴露其激酶结构域,从而激活 CHK1。激活的CHK1被运输到卵胞质,磷酸化CDC25C的Ser216位点,导致CDK1的Thr14和Tyr15磷酸化而抑制其活性,从而阻止G2/M转换并导致细胞周期停滞。此外,受精卵的晚期原核阶段对应于G2阶段,之后受精卵进入有丝分裂。因此,杂合突变增加了受精卵中CHK1的激酶活性,并通过抑制性CDK1的积累引起原核融合失败和合子停滞,从而诱导受精卵G2/M转换停滞[52]。
4.9 突变影响母源效应基因
卵子发生过程中,母源mRNA不断累积,大多数mRNA被翻译成蛋白并且在卵母细胞减数分裂过程中发挥作用,合子形成后这些母源mRNA逐渐被降解。但是,仍然有一些母源mRNA在合子中发挥功能,参与调控早期胚胎发育过程,被称为母源效应基因。皮质下母源复合体(subcortical maternal complex,SCMC)是由多种母源效应基因编码的母源蛋白组成的复合体,对早期胚胎发育具有重要调控作用。人类SCMC的核心组分NLRP2 (NLR family pyrin domain containing 2)、NLRP5 (NLR family pyrin domain containing 5)、OOEP (oocyte expressed protein)、PADI6 (peptidyl arginine deiminase 6)、TLE6 (TLE family member 6)和KHDC3L (KH domain containing 3 like)共定位于卵子和早期胚胎的皮质部分,调控胞质晶格(cytoplasmic lattices,CPLs)的形成,而CPL是母源核糖体和mRNA储存的位置[62]。其中,TLE6参与调控了纤维型微丝网络的动态变化,在卵子由受精前的不对称减数分裂转变为受精后的对称有丝分裂的过程中起到重要作用。纯合或复杂合突变,破坏了TLE6蛋白的结构,影响了TLE6的表达或磷酸化水平,进而破坏TLE6与SCMC其他成员KHDC3L和OOEP的相互作用,导致受精失败和早期胚胎发育停滞[53]。专一的在不同时期卵子以及早期胚胎中表达,与CPL共同定位,其表达水平随着卵子的成熟显著升高,而到8细胞胚胎时则显著降低,至囊胚时期则几乎检测不到表达。基因检测发现,患者携带有基因纯合或复杂合突变,影响了PADI6蛋白的表达,通过剂量效应抑制早期胚胎的合子基因组激活,最终导致早期胚胎停滞和女性不孕[54]。此外,和的隐性突变导致受精失败或者早期胚胎停滞,不同突变对蛋白功能的破坏程度不同,造成患者的表型差异[55]。隐性突变也被发现导致早期胚胎停滞和复发性葡萄胎[56]。
5 结语与展望
减数分裂是人类卵子形成的关键环节,受到了复杂而精妙的调控。辅助生殖技术使人们能够在体外直观地观察到人类卵母细胞减数分裂的形态学变化,并借此展开生理和病理学研究。随着分子生物学技术的进步,对卵子发生的分子机制研究越来越深入;动物模型、辅助生殖和高通量测序技术的广泛应用使人们对卵母细胞减数分裂缺陷的病理机制有了更为清晰地了解。本综述重点介绍了卵子发生的主要细胞和分子事件,特别是卵母细胞减数分裂阻滞和恢复事件,以及该过程的调控机制。此外,本文列举了一系列干扰重要信号通路进而导致卵母细胞成熟缺陷的致病基因,并且介绍了相关致病机制。
间隙连接是卵母细胞与其卵泡环境之间唯一的通讯方式,因此卵母细胞的发育不仅取决于卵母细胞本身,还取决于卵泡内细胞间的相互通讯和物理接触。目前,PI3K/Akt信号通路、AMH、LH等触发卵泡和卵母细胞发育的确切机制仍不确定,需要进一步研究和探索激素和其他细胞外分子在调节卵泡颗粒细胞和卵母细胞分化中的作用[63]。未来的研究应当更多地关注卵母细胞内的表观遗传变化,例如翻译后修饰和非编码RNA在减数分裂中的作用[64]。此外,成像技术、高通量测序技术和组学技术的发展能够将卵子发生的关键事件以更清晰的三维甚至时间依赖性四维方式进行展示[65],借此发现更多的卵母细胞特异性蛋白,如卵母细胞表达的旁分泌分子和RNA结合蛋白,进一步阐明卵母细胞减数分裂的生理机制。同时,已发现的致病基因只能解释少部分不孕不育患者的病因,大多数患者背后的遗传因素和致病机制仍有待发现。因此,卵子发生的生理和病理研究仍然是未来研究的重要课题。
[1] Sen A, Caiazza F. Oocyte maturation: a story of arrest and release., 2013, 5(2): 451–477.
[2] He MN, Zhang T, Yang Y, Wang C. Mechanisms of oocyte maturation and related epigenetic regulation., 2021, 9: 654028.
[3] Pei ZL, Deng K, Xu CJ, Zhang S. The molecular regulatory mechanisms of meiotic arrest and resumption in oocyte development and maturation., 2023, 21(1): 90.
[4] Sang Q, Zhou Z, Mu J, Wang L. Genetic factors as potential molecular markers of human oocyte and embryo quality., 2021, 38(5): 993–1002.
[5] Baerwald AR, Adams GP, Pierson RA. Ovarian antral folliculogenesis during the human menstrual cycle: a review., 2012, 18(1): 73–91.
[6] Orisaka M, Miyazaki Y, Shirafuji A, Tamamura C, Tsuyoshi H, Tsang BK, Yoshida Y. The role of pituitary gonadotropins and intraovarian regulators in follicle development: a mini-review., 2021, 20(2): 169–175.
[7] Zhao Y, Feng HW, Zhang YH, Zhang JV, Wang XH, Liu DT, Wang TR, Li RHW, Ng EHY, Yeung WSB, Rodriguez-Wallberg KA, Liu K. Current understandings of core pathways for the activation of mammalian primordial follicles., 2021, 10(6): 1491.
[8] Rienzi L, Balaban B, Ebner T, Mandelbaum J. The oocyte., 2012, 27(Suppl 1): i2–i21.
[9] Gougeon A. Human ovarian follicular development: from activation of resting follicles to preovulatory maturation., 2010, 71(3): 132–143.
[10] Suarez SS. Mammalian sperm interactions with the female reproductive tract., 2016, 363(1): 185–194.
[11] Sanders JR, Swann K. Molecular triggers of egg activation at fertilization in mammals., 2016, 152(2): R41–R50.
[12] Ahmed TA, Ahmed SM, El-Gammal Z, Shouman S, Ahmed A, Mansour R, El-Badri N. Oocyte aging: the role of cellular and environmental factors and impact on female fertility., 2020, 1247(8): 109–123.
[13] Hunter N. Meiotic recombination: the essence of heredity., 2015, 7(12): a016618.
[14] de Massy B. Initiation of meiotic recombination: how and where? Conservation and specificities among eukaryotes., 2013, 47: 563–599.
[15] Powers NR, Parvanov ED, Baker CL, Walker M, Petkov PM, Paigen K. The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots., 2016, 12(6): e1006146.
[16] Stanzione M, Baumann M, Papanikos F, Dereli I, Lange J, Ramlal A, Tränkner D, Shibuya H, de Massy B, Watanabe Y, Jasin M, Keeney S, Tóth A. Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice., 2016, 18(11): 1208–1220.
[17] Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance., 1999, 19(1): 556–566.
[18] Cloud V, Chan YL, Grubb J, Budke B, Bishop DK. Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis., 2012, 337(6099): 1222–1225.
[19] Adhikari D, Liu K. The regulation of maturation promoting factor during prophase I arrest and meiotic entry in mammalian oocytes., 2014, 382(1): 480–487.
[20] Conti M, Hsieh M, Zamah AM, Oh JS. Novel signaling mechanisms in the ovary during oocyte maturation and ovulation., 2012, 356(1–2): 65–73.
[21] Su YQ, Wigglesworth K, Pendola FL, O'Brien MJ, Eppig JJ. Mitogen-activated protein kinase activity in cumulus cells is essential for gonadotropin-induced oocyte meiotic resumption and cumulus expansion in the mouse., 2002, 143(6): 2221–2232.
[22] Sela-Abramovich S, Chorev E, Galiani D, Dekel N. Mitogen-activated protein kinase mediates luteinizing hormone-induced breakdown of communication and oocyte maturation in rat ovarian follicles., 2005, 146(3): 1236–1244.
[23] Sha QQ, Dai XX, Dang YJ, Tang FC, Liu JP, Zhang YL, Fan HY. A MAPK cascade couples maternal mRNA translation and degradation to meiotic cell cycle progression in mouse oocytes., 2017, 144(3): 452–463.
[24] Wu JQ, Kornbluth S. Across the meiotic divide—CSF activity in the post-Emi2/XErp1 era., 2008, 121(Pt 21): 3509–3514.
[25] Dupré A, Haccard O, Jessus C. Mos in the oocyte: how to use MAPK independently of growth factors and transcription to control meiotic divisions., 2011, 2011: 350412.
[26] Zhang YL, Liu XM, Ji SY, Sha QQ, Zhang J, Fan HY. ERK1/2 activities are dispensable for oocyte growth but are required for meiotic maturation and pronuclear formation in mouse., 2015, 42(9): 477–485.
[27] Krauchunas AR, Wolfner MF. Molecular changes during egg activation., 2013, 102: 267–292.
[28] Sang Q, Ray PF, Wang L. Understanding the genetics of human infertility., 2023, 380(6641): 158–163.
[29] Zhang ZH, Li B, Fu J, Li R, Diao FY, Li CH, Chen BB, Du J, Zhou Z, Mu J, Yan Z, Wu L, Liu S, Wang WJ, Zhao L, Dong J, He L, Liang XZ, Kuang YP, Sun XX, Sang Q, Wang L. Bi-allelic missense pathogenic variants in TRIP13 cause female infertility characterized by oocyte maturation arrest., 2020, 107(1): 15–23.
[30] Wang WJ, Dong J, Chen BB, Du J, Kuang YP, Sun XX, Fu J, Li B, Mu J, Zhang ZH, Zhou Z, Lin Z, Wu L, Yan Z, Mao XY, Li QL, He L, Wang L, Sang Q. Homozygous mutations in REC114 cause female infertility characterised by multiple pronuclei formation and early embryonic arrest., 2020, 57(3): 187–194.
[31] Dong J, Zhang H, Mao XY, Zhu JH, Li D, Fu J, Hu JJ, Wu L, Chen BB, Sun YM, Mu J, Zhang ZH, Sun XX, Zhao L, Wang WJ, Wang WJ, Zhou Z, Zeng Y, Du J, Li QL, He L, Jin L, Kuang YP, Wang L, Sang Q. Novel biallelic mutations in MEI1: expanding the phenotypic spectrum to human embryonic arrest and recurrent implantation failure., 2021, 36(8): 2371–2381.
[32] Christou-Kent M, Kherraf ZE, Amiri-Yekta A, Le Blévec E, Karaouzène T, Conne B, Escoffier J, Assou S, Guttin A, Lambert E, Martinez G, Boguenet M, Fourati Ben Mustapha S, Cedrin Durnerin I, Halouani L, Marrakchi O, Makni M, Latrous H, Kharouf M, Coutton C, Thierry- Mieg N, Nef S, Bottari SP, Zouari R, Issartel JP, Ray PF, Arnoult C. PATL2 is a key actor of oocyte maturation whose invalidation causes infertility in women and mice., 2018, 10(5): e8515.
[33] Chen BB, Zhang ZH, Sun XX, Kuang YP, Mao XY, Wang XQ, Yan Z, Li B, Xu Y, Yu M, Fu J, Mu J, Zhou Z, Li QL, Jin L, He L, Sang Q, Wang L. Biallelic mutations in PATL2 cause female infertility characterized by oocyte maturation arrest., 2017, 101(4): 609–615.
[34] Wu L, Chen H, Li D, Song D, Chen BB, Yan Z, Lyu QF, Wang L, Kuang YP, Li B, Sang Q. Novel mutations in PATL2: expanding the mutational spectrum and corresponding phenotypic variability associated with female infertility., 2019, 64(5): 379–385.
[35] Wang WJ, Guo J, Shi JZ, Li Q, Chen BB, Pan ZQ, Qu RG, Fu J, Shi R, Xue X, Mu J, Zhang ZH, Wu TY, Wang WJ, Zhao L, Li QL, He L, Sun XX, Sang Q, Lin G, Wang L. Bi-allelic pathogenic variants in PABPC1L cause oocyte maturation arrest and female infertility., 2023, 15(6): e17177.
[36] Yang P, Chen TL, Wu KL, Hou ZZ, Zou Y, Li M, Zhang XZ, Xu JT, Zhao H. A homozygous variant in TBPL2 was identified in women with oocyte maturation defects and infertility., 2021, 36(7): 2011–2019.
[37] Huang HL, Lv C, Zhao YC, Li W, He XM, Li P, Sha AG, Tian X, Papasian CJ, Deng HW, Lu GX, Xiao HM. Mutant ZP1 in familial infertility., 2014, 370(13): 1220–1226.
[38] Chen TL, Bian YH, Liu XM, Zhao SG, Wu KL, Yan L, Li M, Yang ZL, Liu HB, Zhao H, Chen ZJ. A recurrent missense mutation in ZP3 causes empty follicle syndrome and female infertility., 2017, 101(3): 459–465.
[39] Dai C, Hu L, Gong F, Tan YQ, Cai SF, Zhang SP, Dai J, Lu CF, Chen J, Chen YZ, Lu GX, Du J, Lin G. ZP2 pathogenic variants cause in vitro fertilization failure and female infertility., 2019, 21(2): 431–440.
[40] Maddirevula S, Coskun S, Al-Qahtani M, Aboyousef O, Alhassan S, Aldeery M, Alkuraya FS. ASTL is mutated in female infertility., 2022, 141(1): 49–54.
[41] Chen BB, Li B, Li D, Yan Z, Mao XY, Xu Y, Mu J, Li QL, Jin L, He L, Kuang YP, Sang Q, Wang L. Novel mutations and structural deletions in TUBB8: expanding mutational and phenotypic spectrum of patients with arrest in oocyte maturation, fertilization or early embryonic development., 2017, 32(2): 457–464.
[42] Feng RZ, Sang Q, Kuang YP, Sun XX, Yan Z, Zhang SZ, Shi JZ, Tian GL, Luchniak A, Fukuda Y, Li B, Yu M, Chen JL, Xu Y, Guo L, Qu RG, Wang XQ, Sun ZG, Liu M, Shi HJ, Wang HY, Feng Y, Shao RJ, Chai RJ, Li QL, Xing QH, Zhang R, Nogales E, Jin L, He L, Gupta ML, Cowan NJ, Wang L. Mutations in TUBB8 and human oocyte meiotic arrest., 2016, 374(3): 223–232.
[43] Wang WJ, Wang WJ, Xu Y, Shi JZ, Fu J, Chen BB, Mu J, Zhang ZH, Zhao L, Lin J, Du J, Li QL, He L, Jin L, Sun XX, Wang L, Sang Q. FBXO43 variants in patients with female infertility characterized by early embryonic arrest., 2021, 36(8): 2392–2402.
[44] Zhao L, Xue SG, Yao ZY, Shi JZ, Chen BB, Wu L, Sun LH, Xu Y, Yan Z, Li B, Mao XY, Fu J, Zhang ZH, Mu J, Wang WJ, Du J, Liu S, Dong J, Wang WJ, Li Q, He L, Jin L, Liang XZ, Kuang YP, Sun XX, Wang L, Sang Q. Biallelic mutations in CDC20 cause female infertility characterized by abnormalities in oocyte maturation and early embryonic development., 2020, 11(12): 921–927.
[45] Zhang YL, Zheng W, Ren PP, Hu HL, Tong XM, Zhang SP, Li X, Wang HC, Jiang JC, Jin JM, Yang WJ, Cao LR, He YL, Ma YR, Zhang YY, Gu YF, Hu L, Luo KL, Gong F, Lu GX, Lin G, Fan HY, Zhang SY. Biallelic mutations in MOS cause female infertility characterized by human early embryonic arrest and fragmentation., 2021, 13(12): e14887.
[46] Zeng Y, Shi JZ, Xu SR, Shi R, Wu TH, Li HY, Xue X, Zhu YC, Chen BB, Sang Q, Wang L. Bi-allelic mutations in MOS cause female infertility characterized by preimplantation embryonic arrest., 2022, 37(3): 612–620.
[47] Zhang YL, Zheng W, Ren PP, Jin JM, Hu ZH, Liu Q, Fan HY, Gong F, Lu GX, Lin G, Zhang SY, Tong XM. Biallelic variants in MOS cause large polar body in oocyte and human female infertility., 2022, 37(8): 1932–1944.
[48] Sang Q, Li B, Kuang YP, Wang XQ, Zhang ZH, Chen BB, Wu L, Lyu QF, Fu YL, Yan Z, Mao XY, Xu Y, Mu J, Li QL, Jin L, He L, Wang L. Homozygous mutations in WEE2 cause fertilization failure and female infertility., 2018, 102(4): 649–657.
[49] Zheng W, Zhou Z, Sha QQ, Niu XL, Sun XX, Shi JZ, Zhao L, Zhang SP, Dai J, Cai SF, Meng F, Hu L, Gong F, Li XR, Fu J, Shi R, Lu GX, Chen BB, Fan HY, Wang L, Lin G, Sang Q. Homozygous mutations in BTG4 cause zygotic cleavage failure and female infertility., 2020, 107(1): 24–33.
[50] Zheng W, Sha QQ, Hu HL, Meng F, Zhou QW, Chen XQ, Zhang SP, Gu YF, Yan X, Zhao L, Zong YR, Hu L, Gong F, Lu GX, Fan HY, Lin G. Biallelic variants in ZFP36L2 cause female infertility characterised by recurrent preimplantation embryo arrest., 2022, 59(9): 850–857.
[51] Wang WJ, Miyamoto Y, Chen BB, Shi JZ, Diao FY, Zheng W, Li Q, Yu L, Li L, Xu Y, Wu L, Mao XY, Fu J, Li B, Yan Z, Shi R, Xue X, Mu J, Zhang ZH, Wu TY, Zhao L, Wang WJ, Zhou Z, Dong J, Li QL, Jin L, He L, Sun XX, Lin G, Kuang YP, Wang L, Sang Q. Karyopherin α deficiency contributes to human preimplantation embryo arrest., 2023, 133(2): e159951.
[52] Zhang HH, Chen TL, Wu KL, Hou ZZ, Zhao SG, Zhang CX, Gao Y, Gao M, Chen ZJ, Zhao H. Dominant mutations in CHK1 cause pronuclear fusion failure and zygote arrest that can be rescued by CHK1 inhibitor., 2021, 31(7): 814–817.
[53] Alazami AM, Awad SM, Coskun S, Al-Hassan S, Hijazi H, Abdulwahab FM, Poizat C, Alkuraya FS. TLE6 mutation causes the earliest known human embryonic lethality., 2015, 16: 240.
[54] Xu Y, Shi YL, Fu J, Yu M, Feng RZ, Sang Q, Liang B, Chen BB, Qu RG, Li B, Yan Z, Mao XY, Kuang YP, Jin L, He L, Sun XX, Wang L. Mutations in PADI6 cause female infertility characterized by early embryonic arrest., 2016, 99(3): 744–752.
[55] Mu J, Wang WJ, Chen BB, Wu L, Li B, Mao XY, Zhang ZH, Fu J, Kuang YP, Sun XX, Li QL, Jin L, He L, Sang Q, Wang L. Mutations in NLRP2 and NLRP5 cause female infertility characterised by early embryonic arrest., 2019, 56(7): 471–480.
[56] Zhang WD, Chen ZL, Zhang DF, Zhao B, Liu L, Xie ZY, Yao YG, Zheng P. KHDC3L mutation causes recurrent pregnancy loss by inducing genomic instability of human early embryonic cells., 2019, 17(10): e3000468.
[57] Jiang ZY, Fan HY. Five questions toward mRNA degradation in oocytes and preimplantation embryos: when, who, to whom, how, and why?, 2022, 107(1): 62–75.
[58] Sha QQ, Zhang J, Fan HY. A story of birth and death: mRNA translation and clearance at the onset of maternal-to-zygotic transition in mammals., 2019, 101(3): 579–590.
[59] Abou-Haila A, Bendahmane M, Tulsiani DR. Significance of egg's zona pellucida glycoproteins in sperm-egg interaction and fertilization., 2014, 66(4): 409–419.
[60] Yu C, Ji SY, Sha QQ, Dang YJ, Zhou JJ, Zhang YL, Liu Y, Wang ZW, Hu BQ, Sun QY, Sun SC, Tang FC, Fan HY. BTG4 is a meiotic cell cycle-coupled maternal-zygotic- transition licensing factor in oocytes., 2016, 23(5): 387–394.
[61] Sha QQ, Yu JL, Guo JX, Dai XX, Jiang JC, Zhang YL, Yu C, Ji SY, Jiang Y, Zhang SY, Shen L, Ou XH, Fan HY. CNOT6L couples the selective degradation of maternal transcripts to meiotic cell cycle progression in mouse oocyte., 2018, 37(24): e99333.
[62] Bebbere D, Albertini DF, Coticchio G, Borini A, Ledda S. The subcortical maternal complex: emerging roles and novel perspectives., 2021, 27(7): gaab043.
[63] da Silveira JC, de Ávila ACFCM, Garrett HL, Bruemmer JE, Winger QA, Bouma GJ. Cell-secreted vesicles containing microRNAs as regulators of gamete maturation., 2018, 236(1): R15–R27.
[64] Eckersley-Maslin MA, Alda-Catalinas C, Reik W. Dynamics of the epigenetic landscape during the maternal-to-zygotic transition., 2018, 19(7): 436–450.
[65] Xu Y, Su GH, Ma D, Xiao Y, Shao ZM, Jiang YZ. Technological advances in cancer immunity: from immunogenomics to single-cell analysis and artificial intelligence., 2021, 6(1): 312.
Physiological and pathological mechanisms of oocyte meiosis
Zhou Zhou1,2, Qing Sang1, Lei Wang1
Normal oogenesis is crucial to successful reproduction. During the human female fetal stage, primordial germ cells transform from mitosis to meiosis. After synapsis and recombination of homologous chromosomes, meiosis is arrested at the diplotene stage of prophase in meiosis I. The maintenance of oocyte meiotic arrest in the follicle is primarily attributed to high cytoplasmic concentrations of cyclic adenosine monophosphate. During the menstrual cycle, follicle-stimulating hormone and luteinizing hormone lead to the resumption of meiosis that occurs in certain oocytes and complete the ovulation process. Anything that disturbs oocyte meiosis may result in failure of oogenesis and seriously affect both the fertilization and embryonic development. The rapid development of the assisted reproduction technology, high-throughput sequencing technology, and molecular biology technology provide new ideas and means for human to understand molecular mechanism of meiosis and diagnosis and treatment of oocyte maturation defects. In this review, we mainly summarize the recent physiological and pathological mechanisms of oogenesis, involving homologous recombination, meiotic arrest and resumption, maternal mRNA degradation, post-translational regulation, zona pellucida assembly, and so on. We wish to take this opportunity to raise the awareness of researchers in related fields on oocyte meiosis, providing a theoretical basis for further research and disease treatments.
oogenesis; oocyte; meiosis; variant
2023-08-07;
2023-10-11;
2023-10-24
国家自然科学基金项目(编号:82288102,32130029,81725006,82171643,81971450),国家重点研发计划(编号:2021YFC2700100)和中国博士后科学基金(编号:2022M712147)资助[Supported by the National Natural Science Foundation of China (Nos. 82288102, 32130029, 81725006, 82171643, 81971450), the National Key Research and Development Program of China (No. 2021YFC2700100) and the China Postdoctoral Science Fund (No. 2022M712147)]
周舟,博士,研究方向:女性生殖遗传学。E-mail: zhouzhoustudy@163.com
王磊,博士,教授,研究方向:生殖遗传学。E-mail: wangleiwanglei@fudan.edu.cn
桑庆,博士,研究员,研究方向:生殖遗传学。E-mail: sangqing@fudan.edu.cn
10.16288/j.yczz.23-170

王磊,教授,博士生导师,国家杰出青年科学基金获得者,国家重点研发计划首席科学家(2021),获科学探索奖(2023)、中国青年科技奖特别奖(2022)、谈家桢生命科学创新奖(2019)等。研究方向为生殖遗传学,重点关注人类卵子、受精及早期胚胎发育的生理与病理机制。以通讯作者在、、、、、等发表多篇论文。揭示了人卵母细胞启动纺锤体组装的独特生理机制(, 2022);发现了首个基因突变导致人类卵子成熟障碍并揭示了致病机制(, 2016),杂志同期配发了编者按,认为这是认识卵子成熟障碍机理迈出的第一步;陆续发现了卵子及胚胎发育异常的4种新遗传病,16个致病基因(国际国内已知致病基因为24个),明确了致病机制并探索了干预策略(, 2023;, 2019;, 2016, 2017, 2018, 2020)。所发现的系列新遗传病及致病基因被国际人类孟德尔疾病数据库OMIM收录,相应基因作为分子指标已被国际国内用于临床患者的疾病诊断。主持国家重点研发计划(项目首席)、国家自然科学基金重点项目和面上项目等。
(责任编委: 史庆华)
猜你喜欢
数学小灵通·3-4年级(2022年10期)2022-12-31
畜牧兽医科技信息(2021年5期)2021-03-05
祝您健康·文摘版(2018年3期)2018-04-06
畜牧兽医科学(2018年12期)2018-02-18
畜牧兽医科学(2018年20期)2018-02-15
畜牧兽医科技信息(2018年5期)2018-02-13
小学生学习指导(低年级)(2017年3期)2017-09-11
时代人物(2015年8期)2015-09-15
恋爱婚姻家庭·养生版(2015年10期)2015-05-14
发明与创新(2015年33期)2015-02-27