
BSA-Seq 结合RNA-Seq 技术挖掘大豆叶片提前黄化衰老基因
2024-01-22 06:44李世宽洪慧龙付佳祺谷勇哲孙如建邱丽娟
作物学报 2024年2期
李世宽 洪慧龙 付佳祺 谷勇哲 孙如建 邱丽娟,*
1 哈尔滨师范大学生命科学与技术学院, 黑龙江哈尔滨 150025; 2 中国农业科学院作物科学研究所, 北京 100081; 3 呼伦贝尔市农业科学研究所, 内蒙古扎兰屯 162650
叶片光合作用是植物生长发育的重要过程, 是农作物产量构成因素之一。叶片变黄是植物开始衰老的显著特征并导致作物减产[1-5]。叶色黄化突变体相关研究始于1933 年, 陆地棉黄叶突变体[6]导致棉花减产, 甚至使棉花死亡。1948 年, Huang 等[7]利用X 射线诱变获得小球藻失绿突变体wcm60, 验证了原卟啉是叶绿素合成的前体, 叶色变异开始逐渐得到重视。
大豆叶片黄化衰老是物质从叶片向发育种子移动的结果[8]。大豆粒重绝大部分来源于开花后的光合产物[9]。在大豆籽粒形成期, 供应中心是本叶腋的籽粒[10]。鼓粒期叶片的光合作用对籽粒生长发育影响最大、其次是荚、再次是叶柄[11], 因此叶片提前黄化衰老会影响大豆产量。1997 年, Lin 等[12]利用Pride B216 × A15 和Anoka × A7 群体定位了缺铁性黄化的7 个QTL, 揭示了缺铁性黄化的多基因控制现象。
Palmer 等发现了非致死型黄化突变体T323, 及其等位的致死型黄化突变体T362H与T225H。用SSR 分子标记对Minsoy ×T225H群体作图, 定位了致死型黄化表型的基因座y18_1与y18_2[13-14]。利用‘波高’ב南农94-156’的重组自交系将与大豆叶绿素累积量相关联的4 个QTL 分别定位于D1a、H、M、G 和F 连锁群[15]。近些年来也不断有报道利用突变体和图位克隆等方法, 将叶片黄化基因定位在1 号、2 号、5 号、10 号、11 号、13 号、15 号、18 号和20 号染色体[16-25], 广泛存在于多条染色体上, 这说明导致叶片黄化的因素有很多。
基因挖掘技术不断创新, 突变体是进行正向遗传学研究的优异材料, 对随机诱变产生的突变体进行研究, 不仅能够挖掘新的基因, 也可以揭示已知基因的新功能[26-29]。混合分组分析法(Bulked Segregant analysis sequencing, BSA-Seq)在挖掘新基因研究中的应用日益广泛[29-33], 该方法在不构建遗传图谱的情况下也能高效快速地挖掘目的基因。处理海量的RNA 测序(RNA sequencing, RNA-Seq)数据,挖掘出有用的信息逐渐成为了研究人员新目标。RNA-Seq 不仅能检测基因表达量, 还可以检测基因的等位变异, 是降低基因组复杂性的一种经济有效的方法。虽然基于RNA-Seq 的SNP/InDel 挖掘只能检测来自转录区域的变异信息, 但是大多数的来自转录区域的SNP/InDel 才是直接影响功能的变异,并且基于RNA-Seq 挖掘特定区域(如转录区域)的SNP/InDel 能提供非常高深度的变异信息, 还能检测到高表达基因中低频的变异[34-35]。竞争性等位基因特异性PCR (kompetitive allelespecific PCR, KASP)技术是基于PCR 的高通量SNP 分型技术, 可基于特定群体的变异信息, 在目标区域开发特异标记, 将图位克隆与KASP 标记的连锁分析相结合的策略,在作物基因挖掘研究中已得到应用[36-38]。
关于全生育期或营养生长期的叶色突变体报道较多, 而在大豆鼓粒后期出现叶片提前黄化衰老性状尚未见报道。本研究在栽培大豆中品661 突变体库中鉴定出一个大豆鼓粒后期非致死型叶片提前10~20 d 黄化突变体ly。利用BSA-Seq 方法初定位并图位克隆, 结合RNA-Seq 技术确定了4 个SNP 和8 个差异表达候选基因, 为探索大豆鼓粒后期叶片提前黄化衰老图位克隆和衰老分子机制研究提供新的思路。
1 材料与方法
1.1 试验材料
在栽培大豆中品661 的EMS 诱变突变体库中鉴定出一个大豆鼓粒后期叶片提前黄化突变体ly。利用ly与大豆鼓粒后期叶片正常色的野生型ofc配制组合得到F0, 在内蒙古扎兰屯市农牧科学研究所温室繁种, 获得F1单株。在北京顺义种植F2获得978个单株, 2020 年种植 F2:3, 根据 F2:3表型取样做BSA-Seq, F2:4取样测RNA-Seq, 2022 年每株F2分出50 粒种子为株行, 海南调查F2:3, 鼓粒后期分别观察F2:3株行表型并取样。
1.2 表型分析
1.2.1 遗传分析 观察F2:3鼓粒后期每行的叶色, 将其分为黄色纯合行、绿色纯合行和分离杂合行, 其中低于30 株的株行表型数据不计入。用适合性测验(χ2测验)检测F2:3黄纯合行、绿纯合行和分离行的行数比。假定无效假设为: F2:3群体的行数比符合1∶2∶1, 若χ2<5.99, 则P>0.05 接受无效假设, 若χ2>5.99, 则P<0.05 否定无效假设。计算公式如下:
式中,O为实际行数;E为理论行数。
1.2.2 生理指标测定 在F2:3鼓粒后期分离行中,分别选择野生型和突变型表型的材料, 取鼓粒充分的3 粒荚, 每组取5 个生物重复。为排除时间和种植位置的影响, 在不同时间和株行分别进行8 次重复。称荚果重和种子鲜重, 种子风干后称干重, 用(种子鲜重-种子干重)/荚果重来表示水分, 种子干重/荚果重来表示干重, 其中荚果重用来消除荚的生长差异, 对得到的数据进行配对t检验, 若P<0.05则代表水分或干重有显著差异; 在表型出现前大约1 周开始用SPAD-502 测定叶片叶绿素含量的相对值即SPAD 值, 直到叶片凋落, 每个性状测3 株, 测3个不同位置叶片, 每个叶片测3 次SPAD 值取平均后计数, 对不同表型得到的数据去掉最大值和最小值后取平均, 并对不同时间的SPAD 值进行t检验若P<0.05 则代表此时的SPAD 值有显著差异。
1.2.3 农艺性状数据分析 在海南三亚冬季短日照条件下大田种植F2:3, 由于野生型亲本ofc(矮皱叶, 图1-A)和突变型亲本ly(高展叶, 图1-C)有明显不同, 因此调查时分为矮皱叶和高展叶2 组, 且选择生长均匀且长势相近的纯合行, 挑选F2:3中2 组的野生型和突变型纯合行各10 行, 每行各10 株完整植株进行考种, 在数据分析时删除相应性状的最大值和最小值, 取平均值进行t检验, 若P<0.05 则代表此性状有显著差异。在北京顺义夏季长日照条件下大田种植, 黄化性状与短日照相同, 因此进行同样的考种和数据分析, 并对2 种条件下获得的数据进行对比测验。
1.3 BSA-Seq 候选区间定位方法
根据2 个亲本及F2:3叶片表型, 构建2 个亲本和2 个叶色差异表型混池, 亲本池分别为10 株野生型(ofc)与10 株突变型(ly); 子代池在F2:3植株中,选择不同黄叶纯合行每行各选1 株, 混合51 单株个体构建突变型混池, 不同绿叶纯合行每行各选1 株,混合48 单株个体构建野生型混池。具体操作步骤如下: 首先, 取植株顶端幼叶, 利用CTAB 法分别提取单株叶片DNA; 检测DNA 质量和浓度, 将不同池的植株DNA 等量混合构建出4 个混池。由北京百迈克生物科技有限公司测序。利用 Illunima Casava 1.8 进行碱基识别分析, 采用双端150 bp 测序策略进行基因组测序。亲本池测序深度为10×, 子代混池测序深度为50×。参考基因组为Wm82.a2.v1版本的大豆基因组[39]。使用GATK[40]软件工具来检测SNP, 利用SnpEff[41]软件进行变异注释和预测变异影响。
1.3.1 ED 关联分析 欧式距离(Euclidean Distance, ED)算法, 是利用测序数据寻找混池间存在显著差异标记, 并以此评估与性状关联区域的方法。理论上, 野生型混池和突变型混池之间除了目标性状相关位点存在差异, 其他位点均趋向于一致, 因此非目标位点的ED 值应趋向于0, ED 值越大表明该标记在两混池间的差异越大, ED 方法的计算公式如下所示, 式中mut 表示碱基在突变混池中的频率, wt表示碱基在野生型混池中的频率。
1.3.2 index 关联分析 index 是通过混池间的基因型频率差异进行标记关联分析的方法, 寻找混池之间基因型频率的显著差异并用Δindex 统计。计算公式如下所示, 式中aa 为子代的野生池即绿叶混池,ab 代表子代的突变池即黄叶混池, M 和P 分别为野生型亲本的等位基因和突变型亲本的等位基因在各自池中出现的read 数目。通过Δindex 可以观察每个位点在突变型和野生型混池之间的差异。标记与性状关联度越强, Δindex 越接近于1。
为检测SNP 和InDel 的可靠性, 对相应位点进行验证。首先在Soybase (http://www.soybase.org/)数据库中查找参考基因组中的变异位点上下游特异序列, 通过DNAMAN 软件(https://www.lynnon.com/)设计引物, 对PCR 扩增产物进行测序, 鉴定目标位点是否存在。引物设置如表1。

表1 7 个变异位点上下游引物序列Table 1 Upstream and downstream primer sequences of 7 mutation sites
1.4 图位克隆
检测SNP/InDel 位点准确性后, 开发分子标记。采用CTAB 法提取对应单株叶片的DNA, 以在F2:3群体中的叶色为表型开展基因型检测, 根据基因型和表型的关系进行图位克隆, 再将标记的基因型与表型进行单标记卡方检测分析, 分析公式为
式中,O为表型与基因型相同的绿叶或黄叶行数;E为鉴定出基因型的绿叶或黄叶行数
1.4.1 竞争性等位基因特异性PCR (kompetitive allelespecific PCR, KASP) 每个标记设计2 条SNP 特异性引物(FAM/HEX)和一条通用引物Rev,FAM 尾部添加能够与FAM 荧光结合的特异性序列(5′-GAAGGTGACCAAGTTCATGCT-3′), F-HEX 尾部添加能够与 H E X 荧光结合的特异性序列(5′-GAAGGTCGGAGTCAACGGATT-3′), 引物序列见表2, KASP 扩增反应体系为10 μL, 具体为: 5 μL 2 × KASP Master Mix, 0.14 μL KASP Primer Mix, 1.0 μL 模板DNA (4~50 ng μL–1), 3.86 μL ddH2O。反应程序为: 95℃热处理10 min; 95℃变性20 s, 61~55℃退火和延伸60 s, 10 个循环(每循环降低0.6℃); 95℃变性20 s, 55℃退火和延伸40 s, 28 个循环; 4℃避光保存, 经荧光信号采集后即可基因分型。

表2 定位区间内7 个标记的引物序列Table 2 Primer sequences of seven markers located in the interval
1.4.2 衍生的酶切扩增多态性标记(derived cleaved amplified polymorphic sequence, dCAPS) 选择在InDel 位点(AGA/A)处设计 dCAPS 标记, 引物为241500D-F 和241500D-MobI-R2, 命名为241500D。下游引物241500D-MboI-R2 序列中引入1 个错配碱基G, 使其在该位点形成了MboI (GATC)的酶切位点。在绿色基因型(caaAGAta)材料中, PCR 扩增产物可被MboI 酶切成155 bp 和28 bp 片段, 黄色基因型(caaAta)材料中, 因不含有MboI 酶切位点而不能被酶切。对PCR 扩增产物酶切, 经聚丙烯酰胺凝胶电泳即可基因分型。
1.5 RNA-Seq 差异基因分析方法
根据2 个亲本及F3:4叶片表型建池, 在鼓粒后期取顶部新鲜叶片, 亲本池分别为10 株野生型(ofc)与10 株突变型(ly); 在F3:4群体中, 子代池选择纯合行黄叶10 个单株构建突变型混池, 绿叶10 个单株构建野生型混池。利用TRIzol Kit 提取叶片总RNA, 采用Truseq RNA 试剂盒进行mRNA 纯化并构建cDNA文库, 对获得的cDNA 文库进行PCR 扩增富集, 采用分子生物学先进设备检测RNA 样品的纯度、浓度和完整性, 以保障使用合格的样品进行转录组测序。库检合格后, 不同文库按照目标下机数据量进行pooling, 用Illumina 平台进行测序。RNA 提取和测序由百迈克生物科技有限公司完成。经过质量控制之后得到的高质量的Clean Data。再利用Tophat将Clean reads 比对到大豆参考基因组, 允许有2 个碱基的错配[42-43]。
1.5.1 变异位点分析 基于各样品 reads 与参考基因组序列的Hisat2 比对结果, 使用GATK[45]软件识别测序样品与参考基因组间的单碱基错配, 识别潜在的 SNP 或InDel 位点。在不同叶色池间筛选变异位点, 再在相同叶色池间确定相同的变异位点,最终获得可靠的变异位点。
1.5.2 差异表达分析 差异表达分析得到的基因集合叫做差异表达基因集, 使用“A_vs_B”的方式命名。根据两组样品之间表达水平的相对高低,差异表达基因划分为上调基因(up-regulated gene)和下调基因(down-regulated gene)。上调基因在样品(组) A 中的表达水平高于样品(组) B 中的表达水平;反之为下调基因, 无表达差异基因为(no-differential genes)。表达定量的结果以FPKM(fragments per kilobase ofexon model per million mapped fragments) 为单位, 在得到差异检验的FDR 值同时, 根据基因的表达量(FPKM 值)计算该基因在不同样本间的差异表达倍数。以“FDR≤0.001 和|log2Ratio (Fold Change)|≥1”作为阈值来判断基因差异表达的显著性[44]。根据不同池得到的差异表达基因, 在野生型亲本vs 突变型亲本、野生型子代vs 突变型子代、野生型亲本vs 突变型子代、突变型亲本 vs 野生型子代中筛选差异表达基因,这4 个结果取交集得到不同叶色间差异基因; 在野生型亲本vs 野生型子代、突变型亲本vs 突变型子中的差异表达基因, 这2 个结果都不是不同叶色间的差异表达基因, 因此应取并集, 并在取交集得到的基因中排除, 即得到野生型和突变型间准确的差异表达基因。
2 结果与分析
2.1 大豆鼓粒后期叶片黄化性状分析
2.1.1 遗传分析 凤交66-12、冀豆12、郑1307和ofc分别与ly杂交, F1叶色表现为绿色, 表明此性状为隐性性状。ofc×ly群体F2:3的大豆鼓粒后期叶色的绿色纯合行、黄色纯合行和杂合行行数分别为212、203 和418, 对其进行适合性测验, χ2(1:2:1)=0.213< χ20.05,2=5.99,P=0.9, 说明性状符合1∶2∶1 的概率为90%, 黄化性状由1 对隐性基因控制。
2.1.2 黄化性状的变化特点 观察F2:3鼓粒后期不同植株、叶、荚和种子的黄化性状, 从表型开始出现差异到叶片凋落, 颜色有明显差异; 其中突变型为叶脉间开始失绿变黄到整个植株失绿变黄, 而后叶片褐变凋落, 野生型为正常变黄凋落(图2-A),此表型是一个渐变的过程, 突变型出现黄色表型早;从衰老情况看, 突变型叶片更容易褐化凋落(图2-B);从突变型的成熟荚和种子上看, 突变型比野生型发白(图2-C, D)。


图2 野生型与突变型鼓粒后期植株整体的差异变化(A)、叶色变化差异(B)、荚色变化差异(C)和成熟种子的差异(D)Fig.2 Differences in overall changes of plants in soybean filling later period of wild type and mutant tympanum (A), leaf color change (B), pod color change (C), and mature seeds (D)
大豆鼓粒后期水分和干物质的测定结果表示(表3), 突变型与野生型水分差异不明显, 但突变型的干重较高, 结合植株成熟后的考种数据, 认为此性状使鼓粒时的干物质提前积累。
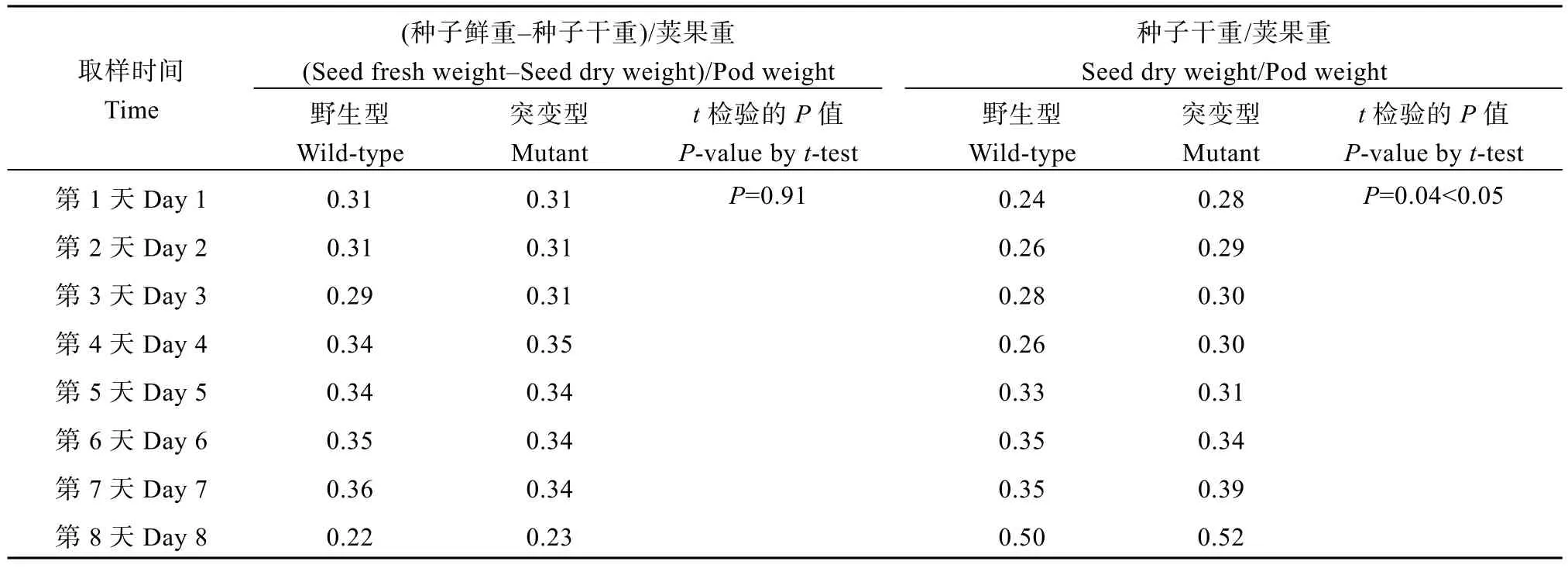
表3 突变型和野生型鼓粒后期水分和干物质的差异Table 3 Differences in moisture and dry matter at the late stages of mutant and wild tympanums
对大豆鼓粒后期的叶片SPAD 值进行分析(图3)后, 发现至表型出现后随着时间的推移, 二者SPAD值差异越发明显, 直到两表型均测不出SPAD 值为止, 二者的SPAD 值均有明显差异, 说明叶绿素含量在二者间存在显著差异。
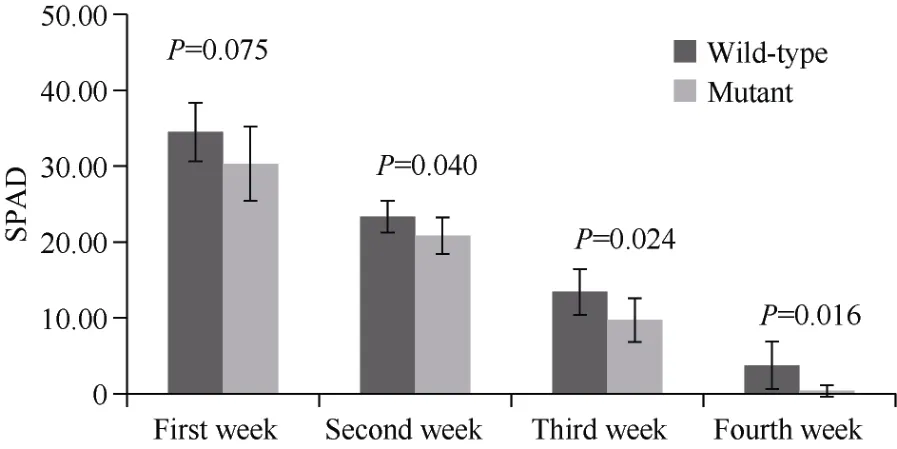
图3 大豆鼓粒后期叶片SPAD 值的变化Fig.3 Changes of SPAD values of leaves at the late stage of soybean drum grain
2.1.3 其他农艺性状比较 对矮皱叶和高展叶两组的野生型和突变型的考种数据分析(表4)认为短日照条件下差异性状较多, 矮皱叶组突变型株高、荚数、粒数和百粒重都显著低于野生型, 高展叶组突变型株高、荚数、粒数和百粒重也显著低于野生型, 分枝数显著高于野生型; 长日照下差异性状较少, 矮皱叶组只有荚数突变型显著高于野生型, 高展叶组突变型分支和粒数显著高于野生型, 百粒重显著低于野生型。
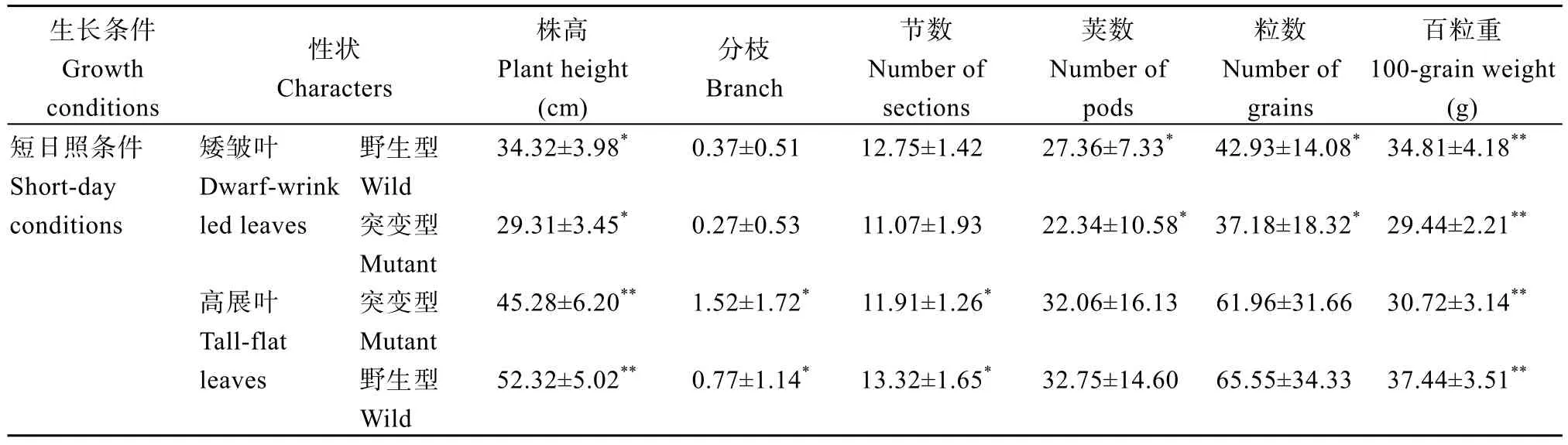
表4 野生型和突变型的考种数据分析Table 4 Analysis of wild-type and mutant-type agronomic trait data
2.2 基于BSA-Seq 筛选黄化性状的定位区间
2.2.1 数据质控 2 个亲本池与2 个子代池通过Illumina HiSeq 重测序, 测序共得到的Clean Data 为141.04 Gbp, Q30 达到90.56。样品与参考基因组平均比对效率为98.70%, 平均覆盖深度为30.50X, 基因组覆盖度为99.33% (至少一个碱基覆盖), 样品AT、CG 碱基基本不发生分离且各插入片段大小的分布呈单峰的正态分布, 说明测序数据质量合格, 与大豆参考基因组比对效率较高, 可用于后续分析。
2.2.2 关联分析 进一步过滤SNP/InDel, 得到高质量可信SNP/InDel 位点12,160/3218 个。采用ED 算法和index 2 种算法对这些高质量SNP/InDel进行关联。利用两混池间基因型存在差异的SNP/InDel 位点, 统计各个碱基在不同混池中的深度, 并计算每个位点ED 值, 为消除背景噪音, 对原始ED 值进行5 次方处理[45], 然后采用DISTANCE方法对 ED 值进行拟合, 取所有位点拟合值的median+3SD 作为分析的关联阈值[46], 计算得0.50/2.09。根据关联阈值判定, SNP/InDel 的ED 关联结果如图4-A 所示, index 关联结果如图4-B 所示。
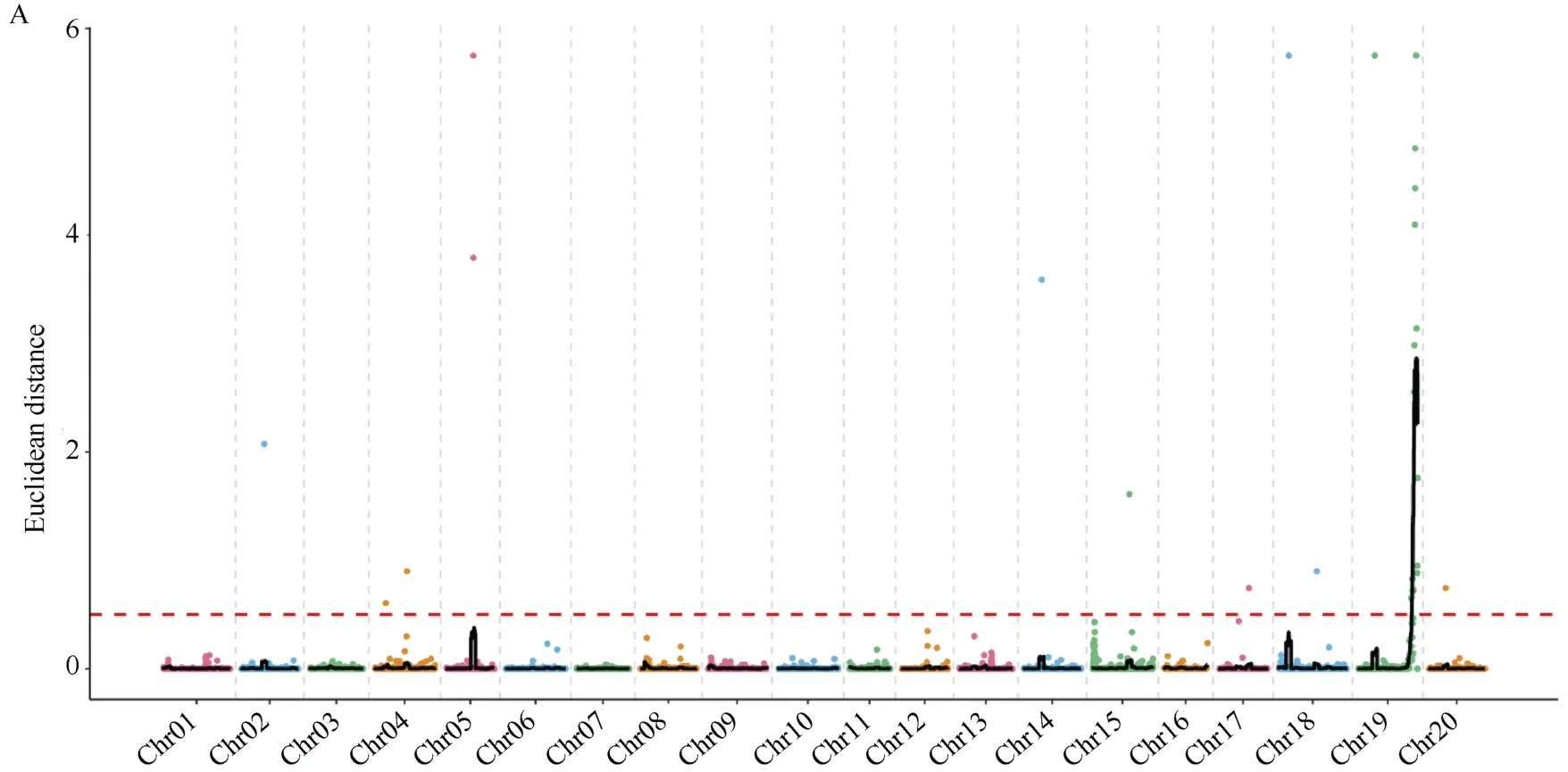
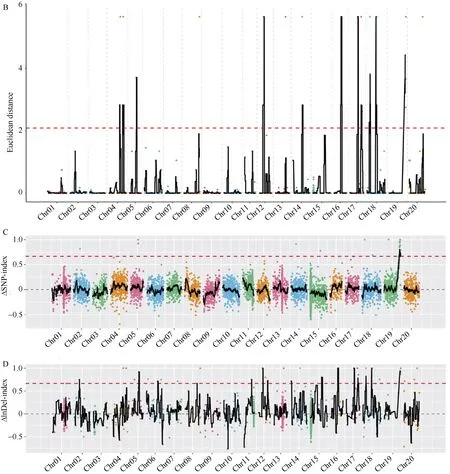
图4 SNP/InDel 的ED 和index 关联分析结果Fig.4 ED and index correlation analysis of SNP/InDel
根据SNP 的关联分析在19 号染色体定位区间4.96 Mb, 包括636 个基因。根据InDel 关联分析定位了包括19 号染色体在内的8 个区间, 2 种关联分析方法得到的结果取交集, 仅有1 个区域(表5), 总长度2.23 Mb, 共包含277 个基因。将测序分析结果和关联分析结果可视化, 并将样品的变异结果及BSA 关联分析结果使用circus 软件(http://circos.ca/)作图(图5)。
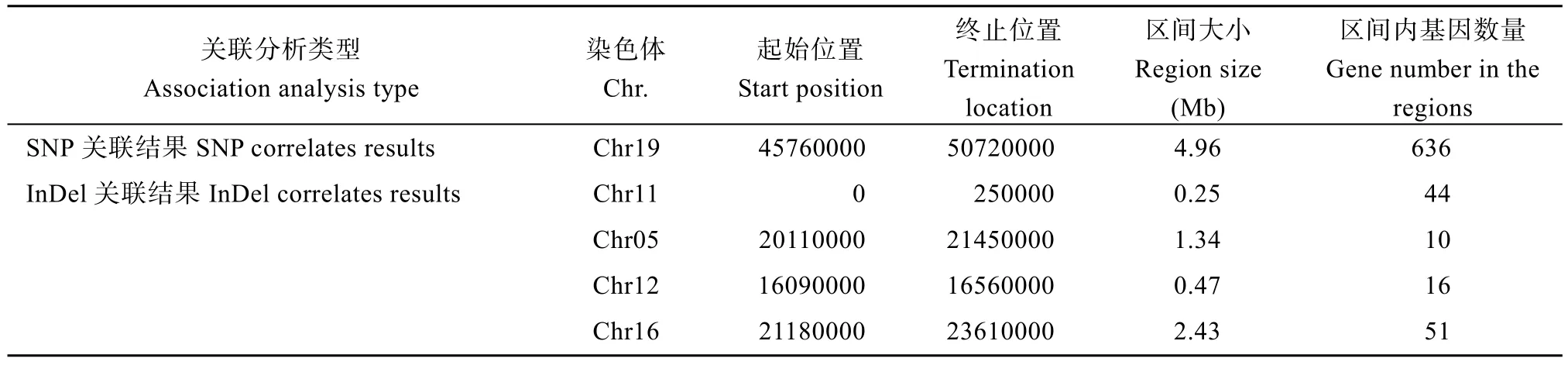
表5 SNP/InDel 分析结果利用2 种方法在19 号染色体获得关联区域Table 5 Associated region on chromosome 19 based on SNP/InDel analysis by two methods
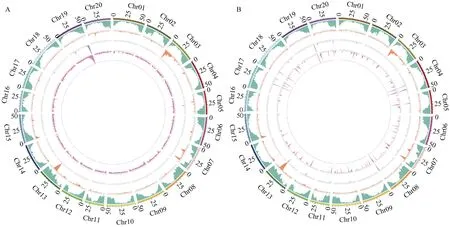
图5 SNP (A)和InDel (B)在染色体上结果的可视化分布Fig.5 Visual distribution of SNP (A) and InDel (B) results on chromosomes
2.3 开发标记加密定位区间
根据BSA-Seq 分析得到的突变位点, 利用已成功开发的6 个Kasp 标记和一个dCaps 标记对F2:3纯合行行混进行标记-表型连锁分析(图6), 将区间从2.23 Mb 缩短为物理位置为47,192,234~48,945,702,一共1.75 Mb, 其中含有219 个基因, 其中有SNP 突变的基因5 个。结合单标记分析方法(表6)结果显示,Glyma.19G223400到Glyma.19G237000之间标记均是χ2< χ20.05,1=3.84, 其中标记237,000 的P值最大,说明在223,400 到237,000 之间的标记基因型与表型相符概率大, 认为其与目标基因的连锁程度最高。
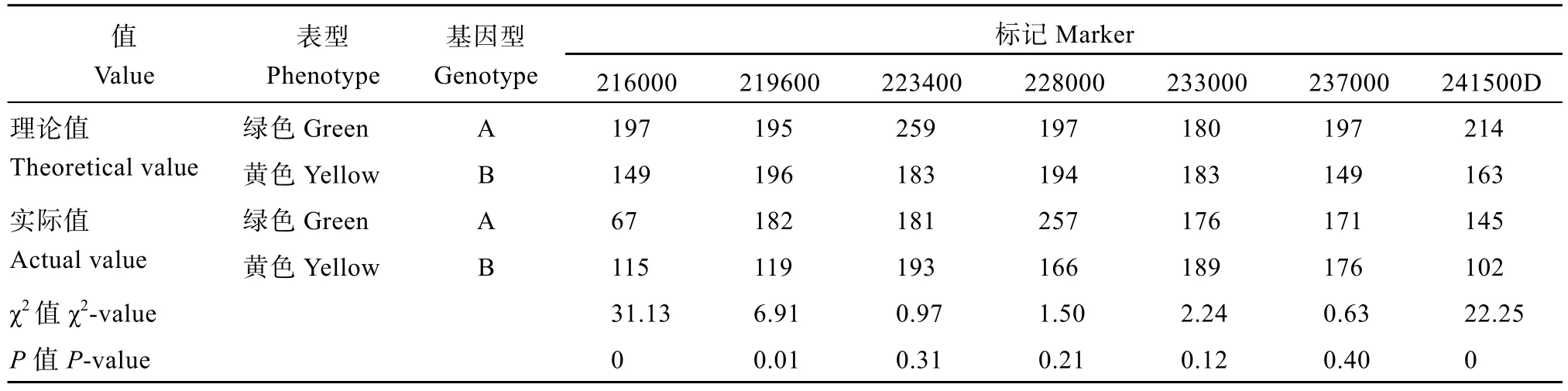
表6 7 个标记在F2:3 纯合行的单标记分析Table 6 Single label analysis of seven markers in F2:3 homozygous rows
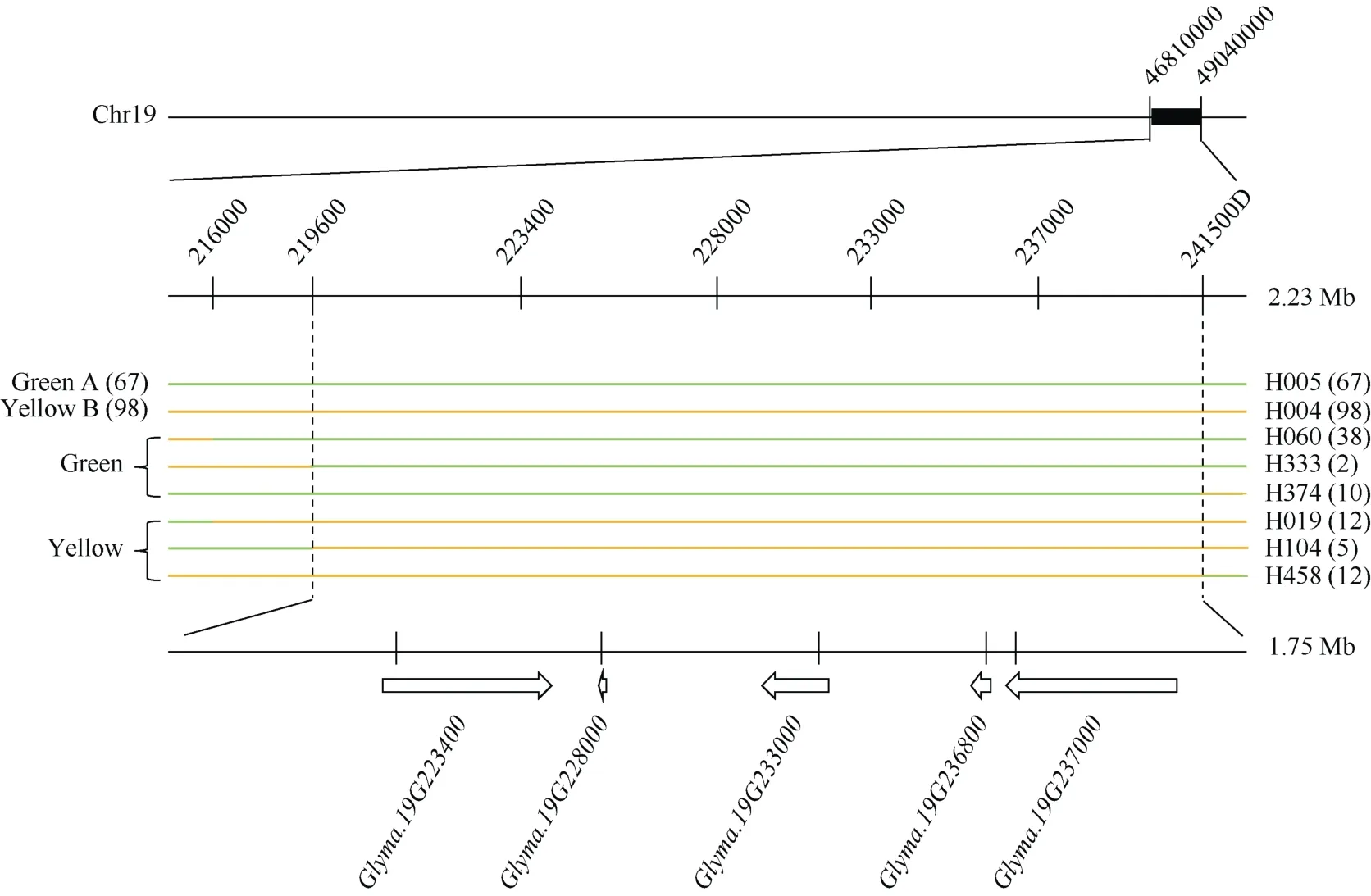
图6 7 个标记在F2:3 纯合行的表型连锁分析Fig.6 Phenotypic linkage analysis of seven labeled homozygous rows at F2:3
2.4 RNA-Seq 差异基因
2.4.1 数据质控 经过测序质量控制, 共得到24.81 Gb Clean Data, 各样品Q30 碱基百分比均不小于 94.94%。样品与参考基因组从比对结果统计来看,各样品的 Reads 与参考基因组的比对效率在52.50%~96.53%之间。通过检验插入片段在基因上的分布, reads 在所有表达的基因上的分布呈现均一化分布, 在插入片段大小的分布呈单峰的正态分布,使用各样品的Mapped Data 对检测到的不同表达情况的基因数目饱和情况进行模拟, 随着测序数据量的增加, 数值越趋近于1, 这说明mRNA 片段化的随机性高、无mRNA 降解情况, 插入片段长度的离散程度高, 文库容量和Mapped Data 充足[47]。可用于后续候选区间的基因筛选。
2.4.2 变异位点分析 对RNA-Seq 得到的变异基因进行筛选, 在已得区间内有4 个SNP 差异的基因(表7 和表8),Glyma.19G223400、Glyma.19G233000和Glyma.19G237000均发生在UTR 区域, 可能导致RNA 二级结构发生改变, 进而影响RNA 的稳定性和翻译效率,Glyma.19G236800则是在CDS 区发生改变, 使第185 位脯氨酸变成亮氨酸, 改变了蛋白的锌指结构域, 从而影响蛋白的功能。
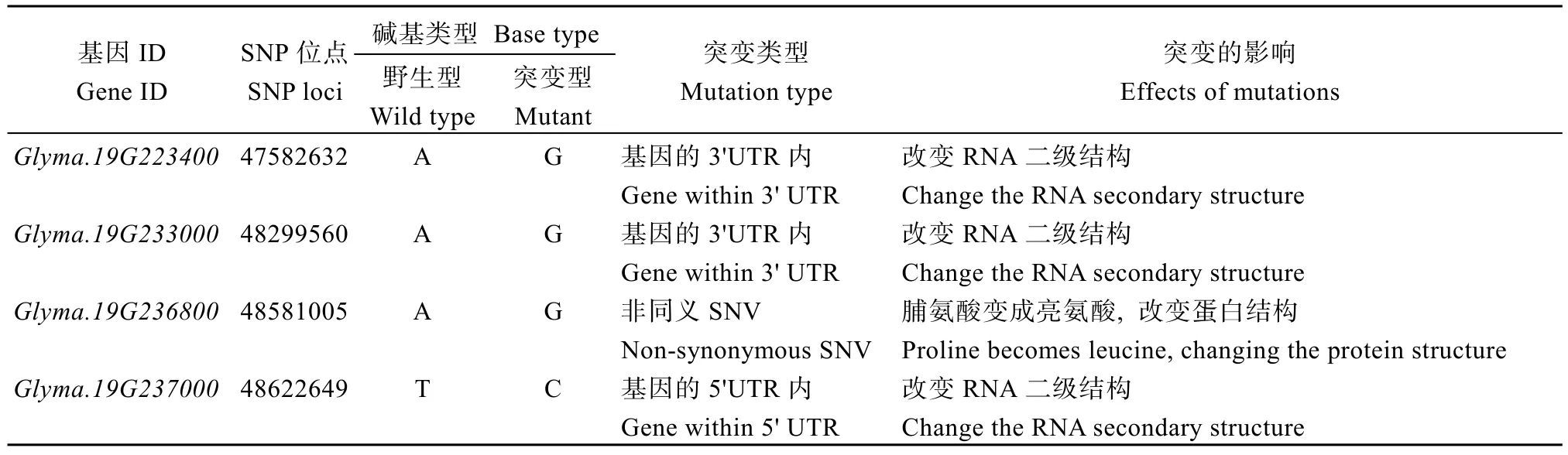
表7 SNP 突变的4 个候选基因Table 7 Four candidate genes for SNP mutations
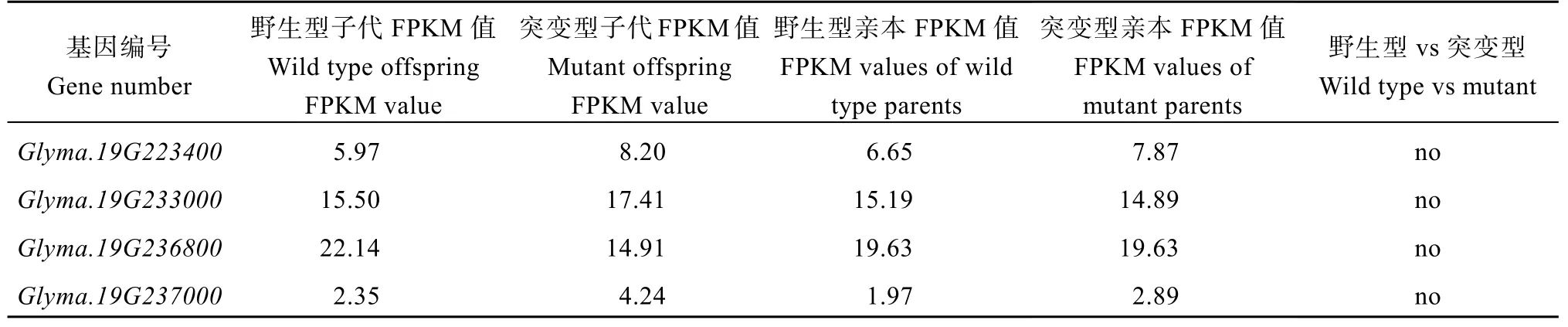
表8 4 个SNP 基因的表达信息Table 8 Relative expression information of four SNP genes
2.4.3 差异表达基因分析 通过不同叶色池间差异表达基因取交集, 得到2948 个基因(图7-A); 相同叶色池间差异表达基因取并集, 得到4780 个基因(图7-B), 而后从取交集的基因中排除取并集中的相同基因, 得到2392 个与大豆鼓粒后期叶色差异相关基因, 这些差异表达基因与获得的区间取交集, 得到8 个基因(表9)。
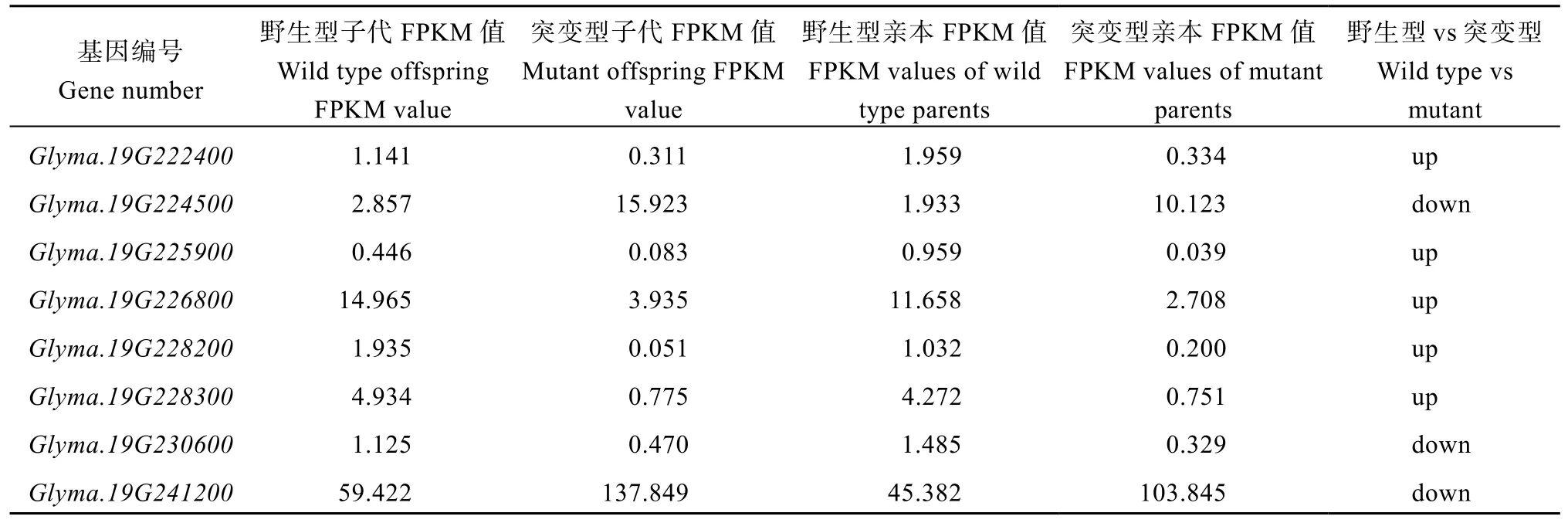
表9 区间内8 个差异表达基因Table 9 Eight differentially expressed genes in the interval
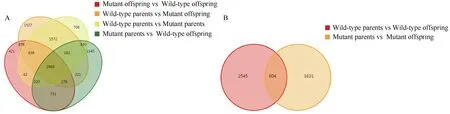
图7 野生型和突变型的亲本或子代间的差异基因取交集(A)亲本和子代的野生型或突变型的差异基因取并集(B)Fig.7 Intersection of differential genes between parents or offspring of wild-type and mutant type (A) combination of differentiating genes of wild-type or mutant type of parents and offspring (B)
2.5 候选基因生物信息学分析
2.5.1 表达谱和蛋白序列分析 为了进一步探究共定位区间内候选基因在各个组织中的表达情况,利用phytozome (/phytozome.jgi.doe.gov/)数据库查询了4 个SNP 差异基因和8 个表达差异基因在Williams 82中的表达数据, 从构建不同部位表达图谱(图8)结合蛋白功能注释(表10)中可以看出,Glyma.19G237000、Glyma.19G225900、Glyma.19G226800和Glyma.19G228200在各部位中的表达量都低, 它们与核苷三磷酸水解和碳水化合物代谢过程相关;Glyma.19G222400、Glyma.19G223400和Glyma.19G233000表达模式相近, 均是花中表达低其他部位表达高, 其与乙酰转移和信号转导有关,Glyma.19G224500与这些基因的表达模式相反, 在花中表达低其他部位高, 同样也与乙酰转移可能有关;Glyma.19G230600在芽顶端分生组织、茎和种子中表达相对较高, 其与蛋白磷酸化有关;Glyma.19G228300在茎和果荚中表达较高,Glyma.19G236800在叶以及根、根瘤和根毛中表达较高, 与泛素化蛋白降解有关;Glyma.19G241200在花、叶、果荚以及芽顶端分生组织中表达较高, 与酰基转移相关。
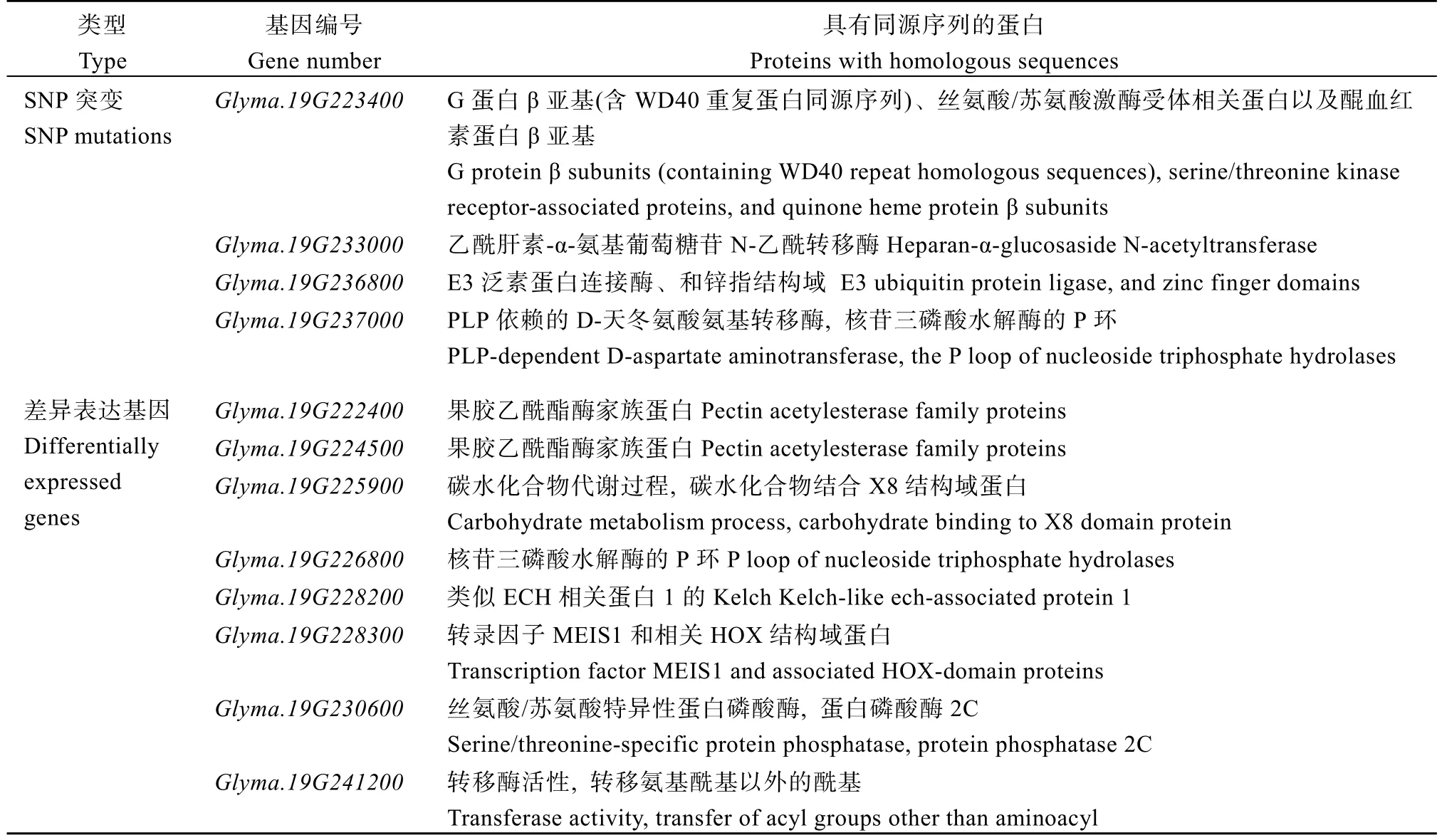
表10 候选基因蛋白序列BLAST 分析Table 10 BLAST analysis of candidate gene protein sequences
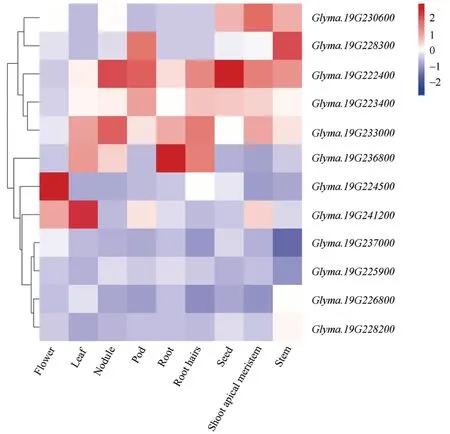
图8 12 个候选基因的表达图谱Fig.8 Relative expression profile of 12 candidate genes
3 讨论
3.1 水分和干物质的测定方法
有试验表明, 利用荚果厚度可以模拟大豆的鼓粒进程[48], 大豆荚果发育过程有2 个部分重叠的阶段: 结荚(建库)阶段和鼓粒(籽粒充实)阶段。荚长和荚宽的增加主要集中在结荚阶段, 荚果长度和宽度达到最大值时大豆的干重只有最终干重的4%[49]。荚果厚度增加略晚于荚长和荚宽, 而与大豆鼓粒相一致[50]。这说明荚果可以在一定程度上反应鼓粒进程, 而本试验的性状在鼓粒后期, 也就是粒鼓满后表型开始发生变化, 因此在试验方法上, 选择使用荚果的鲜重来模拟进程, 可以消除生长上的差异,从而探究水分和干物质的变化。
3.2 大豆鼓粒期干重变化
鼓粒期之前, 豆荚发育从叶片获得较多的光合产物来构建自身, 并通过较强的呼吸作用为其自身发育提供能量。而进入鼓粒期后, 叶片光合能力达到最大[51-53], 在鼓粒前期, 豆荚具有较强的呼吸作用, 呼吸底物除豆荚自身光合形成的光合产物外,主要来源于叶片光合产物, 此时期豆荚的光合产物对籽粒重的贡献较大(贡献率为7.34%~15.06%)。在鼓粒中期(开花26~40 d), 叶片干重先上升后再下降,光合产物主要向籽粒输送, 此时期籽粒干重增加迅速, 鼓粒速度快, 粒重增加也快, 以生殖生长为主;在鼓粒末期(开花40 d 后), 叶片干重下降, 鼓粒速度下降, 但粒重仍在增加, 单株籽粒产量呈上升趋势,营养体生长停止, 生殖生长还在继续[54]; 本实验研究的性状发生在鼓粒后期R5 至R6 期, 也就是在籽粒生殖生长阶段, 此时的SPAD 值说明黄化性状的叶绿素含量下降, 认为在营养体生长停止后野生型的植株依然继续一段生殖生长, 而突变型的植株不再进行生殖生长直接将干物质运往籽粒, 因此导致百粒重上发生显著的差异。虽然有报道认为百粒重与籽粒含水量关系密切, 呈显著负相关[54], 但在本研究中, 鼓粒后期黄化与正常性状的水分差异不明显, 也就说明了黄化性状的叶片在营养体生长停止后, 干物质迅速运往籽粒, 但生殖生长阶段的叶片光合时间变短, 最终运往籽粒的总干物质减少, 导致百粒重下降。因此本研究的黄化性状是牺牲籽粒部分干重为代价, 提前将干物质积累完毕。可以应用于鼓粒后期应对气候变化等未知因素时促使叶片干物质提前转移完毕, 相对提高一些产量。
3.2 候选基因预测
大豆鼓粒期可溶性糖含量的变化是植物体内碳水化合物代谢的重要指标, 既能够反映碳水化合物的合成情况, 也能够说明碳水化合物在植物体内的运输情况[55-57]。大豆鼓粒期叶片和荚皮都是糖代谢相对比较旺盛的部位, 为籽粒运输更多的碳水化合物奠定基础, 可溶性糖含量为叶片>荚皮>茎秆>根系[58]。本试验对RNA-Seq 得到的2392 个叶色差异表达基因进行生物信息学COG 和KOG 的功能分类分析, 发现了4 个碳水化合物的运输和代谢功能相关基因, 为Glyma.19G233000、Glyma.19G222400、Glyma.19G224500和Glyma.19G225900, 它们有可能是大豆鼓粒期后期黄化性状的目的基因; 在KEGG通路富集和KEGG 分类分析中认为与植物激素信号转导相关的候选基因Glyma.19G223400、Glyma.19G236800和Glyma.19G230600也有可能是大豆鼓粒期后期黄化性状的目的基因, GO 富集分析认为跟质膜组分相关, 而Glyma.19G236800也同样是质膜组分, 且4 个SNP 基因分析发现, 3 个UTR 区域突变并没有改变表达量,Glyma.19G236800有着蛋白序列的变异, 因此认为其更可能是黄化性状的目的基因。
4 结论
突变体ly的鼓粒后期黄化性状受单个隐性核基因控制, 基于BSA-Seq、RNA-Seq 和图位克隆将该基因定位在19 号染色体上, 获得1.84 Mb 的区间,区间内有219 个基因, 有SNP 的基因4 个, 有差异表达基因8 个, 它们可能与大豆鼓粒后期叶片黄化衰老性状相关, 但还需后期试验筛选和验证这12 个候选基因。本研究为探索大豆叶片黄化的分子调控机制奠定了基础。
猜你喜欢
广西医学(2020年13期)2020-03-04
商品与质量(2019年36期)2019-04-15
现代园艺(2017年21期)2018-01-03
中国果业信息(2017年4期)2017-05-09
中国果菜(2016年9期)2016-03-01
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
中国现代医生(2015年5期)2015-03-31
现代检验医学杂志(2015年5期)2015-02-06
安徽医药(2014年9期)2014-03-20
