
南五味子药材UPLC指纹图谱及一测多评法研究
2024-01-24 09:33刘晓霞位翠杰冯涌微张芳平段志文谢文凯何民友李振雨
天然产物研究与开发 2024年1期
刘晓霞,位翠杰,冯涌微,张芳平,段志文,谢文凯,何民友,李振雨
广东一方制药有限公司 广东省中药配方颗粒企业重点实验室,佛山 528244
南五味子为木兰科植物华中五味子SchisandrasphenantheraRehd.et Wils.的干燥成熟果实[1]。其性温,味酸、甘,具有收敛固涩、益气生津、补肾宁心的功效,用于久嗽虚喘、梦遗滑精、遗尿尿频、久泻不止、自汗盗汗、津伤口渴、内热消渴、心悸失眠等证。南五味子主要化学成分为三萜类和木脂素类化合物。近年来研究表明南五味子具有保肝护肝、镇静催眠、抗肿瘤、降血糖、抗氧化及增强免疫力等作用,具滋补强壮之力,有很高的药用和食用价值。
中药的质量,直接关系到临床治疗的效果,有效控制中药的质量,是保证中医临床疗效的前提[2]。目前,指纹图谱技术已被广泛地应用到中药质量控制中[3,4],它能较全面地表征中药化学成分,具有特征性和全面性的特点[5],将指纹图谱与一测多评法(QAMS)相结合从定性和定量角度全面控制中药的质量,是中药现代化的重要途径[6]。目前,2020年版《中华人民共和国药典》(以下简称“《中国药典》”)南五味子药材项下以五味子酯甲为含量测定指标,但单一成分的含量测定已不能满足中药整体的质量控制理念。
目前,已有关于南五味子指纹图谱的建立和化学成分的研究报道,如Guo等[7]采用高效液相色谱法(HPLC)建立了南、北五味子药材指纹图谱,结合化学模式识别分析,筛选出了4个影响南、北五味子药材质量的差异标志物。Ma等[8]等采用HPLC法建立南五味子药材指纹图谱,并对指纹图谱中的7个已知成分进行定量分析。Wei等[9]采用UPLC法建立了南五味子药材指纹图谱,并对五味子中的3种主要有效成分五味子甲素、安五脂素和五味子酯甲的含量进行测定。相较于普通液相色谱,超高效液相色谱的使用能够缩短分析时间,提高色谱峰的分离度,在指纹图谱的研究中优势更为明显,而上述研究面临的共同问题是法定对照品的缺失以及对照品的过多使用带来成本的上升,采用一测多评法用于南五味子药材的质量控制研究仍未有报道。本次研究采用UPLC指纹图谱和一测多评,结合化学计量学分析手段,对14批对南五味子药材进行全面质量控制研究,既节省了对照品,又为全面、客观、准确评价南五味子质量提供依据。
1 仪器与材料
1.1 仪器
Themo Vanquish超高效液相色谱仪(赛默飞有限公司);ACQUITY HSS T3 C18色谱柱(2.1 mm×150 mm,1.8 μm);ME204E万分之一天平(梅特勒-托利多公司);XP26百万分之一天平(梅特勒-托利多公司);KQ-500DE数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试剂与试药
五味子酯甲(批号:111529-201706,含量:95.20%)、五味子甲素(批号:110764-201915,含量:99.50%)(中国食品药品检定研究院);五味子酯丙(批号:ST24450105,含量:98.0%)、安五脂素(批号:ST0 7390120,含量:98.0%)(上海诗丹德标准技术服务有限公司);五味子酯乙(批号:18092804,含量:98.0%,成都普菲德生物技术有限公司);五味子酯丁(批号:CFS202002,含量:98.0%,Chem Faces公司);液相用乙腈(色谱纯,默克股份有限公司);水为超纯水(实验室自制);其余试剂皆为分析纯。
1.3 药材
本次实验所用的南五味子药材由广东一方制药有限公司采购管理部采收,经广东一方制药有限公司孙冬梅主任中药师鉴定为木兰科植物华中五味子SchisandrasphenantheraRehd.et Wils.的干燥成熟果实,14批南五味子产地信息见表1。
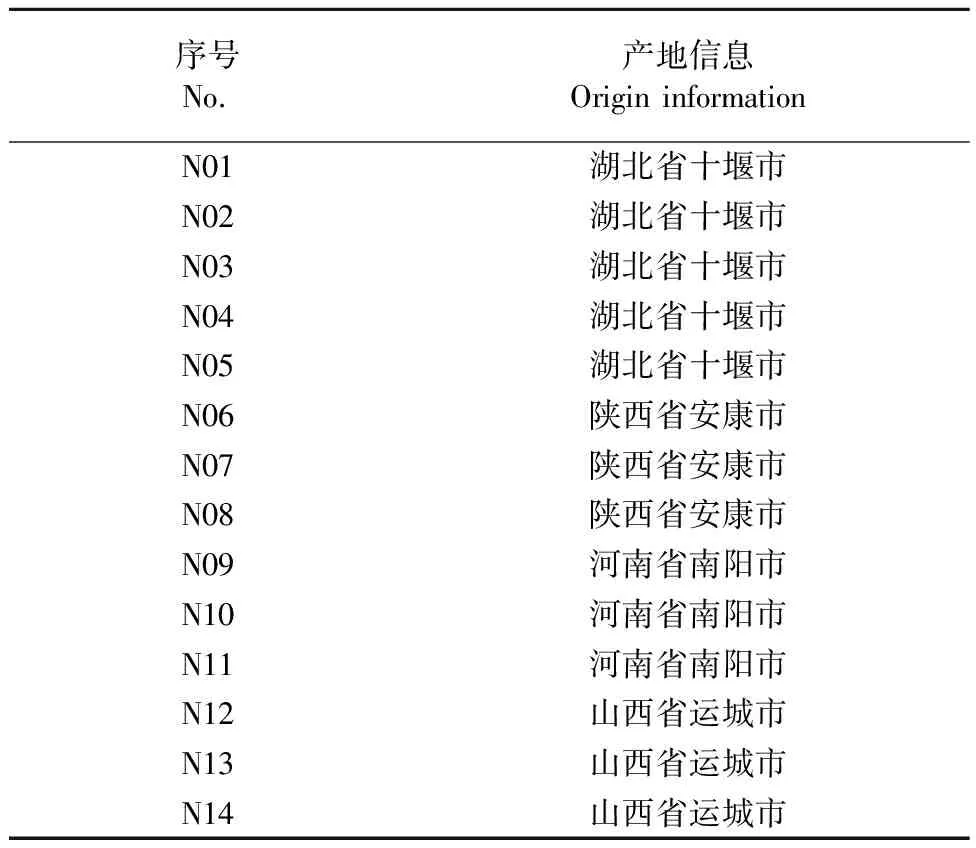
表1 南五味子药材产地信息表
2 方法与结果
2.1 指纹图谱的建立
2.1.1 色谱条件
采用Waters ACQUITY HSS T3(2.1 mm×150 mm,1.8 μm)色谱柱;以乙腈为流动相A,0.1%磷酸溶液为流动相B,梯度洗脱(0~10 min,2%→10%A;10~11 min,10%→51%A;11~26 min,51%→54%A;26~40 min,54%→95%A);流速为0.3 mL/min;柱温为30 ℃;检测波长为230 nm;进样量为1 μL。
2.1.2 对照品溶液的制备
精密称取原儿茶酸对照品适量,加甲醇制成每1 mL原儿茶酸85.926 μg的溶液,作为对照品溶液;另精密称取五味子酯丙、五味子酯甲、五味子酯乙、安五脂素、五味子甲素和五味子酯丁对照品适量,加甲醇制成每1 mL含五味子酯丙58.252 μg、五味子酯甲98.589 μg、五味子酯乙102.116 μg、安五脂素97.334 μg、五味子甲素102.246 μg和五味子酯丁80.634 μg的混合溶液,即得。
2.1.3 供试品溶液的制备
取南五味子药材粉末(过三号筛)约0.5 g,精密称定,置具塞锥形瓶中,精密加入70%乙醇50 mL,称定重量,超声处理(功率250 W,频率40 kHz)30 min,取出,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.1.4 指纹图谱方法学考察
2.1.4.1 精密度试验
取南五味子药材供试品溶液,按“2.1.1”项下色谱条件连续进样6次,以五味子酯甲峰为参照峰S,计算各共有峰与S峰的相对保留时间和相对峰面积及RSD值,相对保留时间RSD值在0.20%~0.96%范围内,相对峰面积RSD值在0.06%~3.0%范围内,表明仪器精密度良好。
2.1.4.2 重复性试验
取同一份样品(编号:N01,过三号筛)约0.5 g,按“2.1.3”项下方法制备6份供试品溶液,按“2.1.1”项下色谱条件测定,以五味子酯甲峰为参照峰S,计算各共有峰与S峰的相对保留时间和相对峰面积及RSD值,相对保留时间RSD值在0.070%~1.3%范围内,相对峰面积RSD值在0.20%~2.1%范围内,表明该方法重复性良好。
2.1.4.3 稳定性试验
取“2.1.4.2重复性试验”项下供试品溶液,按“2.1.1”项下色谱条件,分别在0、2、5、7、12、18、24 h进行分析,以五味子酯甲峰为参照峰S,计算各共有峰与S峰的相对保留时间和相对峰面积及RSD值,相对保留时间RSD值在0.10%~2.2%范围内,相对峰面积RSD值在0.14%~2.3%范围内,表明供试品溶液在24 h内稳定性良好。
2.1.5 指纹图谱的建立及共有峰标定
根据“2.1.1”项下色谱条件和“2.1.3”项下供试品溶液制备方法,对14批南五味子药材进行指纹图谱测定,记录色谱图,导出指纹图谱CDF格式,并导入《中药色谱指纹图谱相似度系统(2012.130723版本)》,以编号N01样品指纹图谱为参照图谱,时间窗宽度设定为0.1 min,进行多点校正和色谱峰匹配,14批南五味子药材指纹图谱共标识出15个共有峰,其叠加图见图1所示。采用中位数法生成南五味子药材对照指纹图谱(见图2)。采用“2.1.3”项下对照品溶液对共有峰进行指认,通过与对照品保留时间和紫外-可见光3D光谱对比分析,确定峰1为原儿茶酸;峰5为五味子酯丙;峰6为五味子酯甲;峰7为五味子酯乙;峰11为安五脂素;峰13为五味子甲素;峰14为五味子酯丁(见图3)。
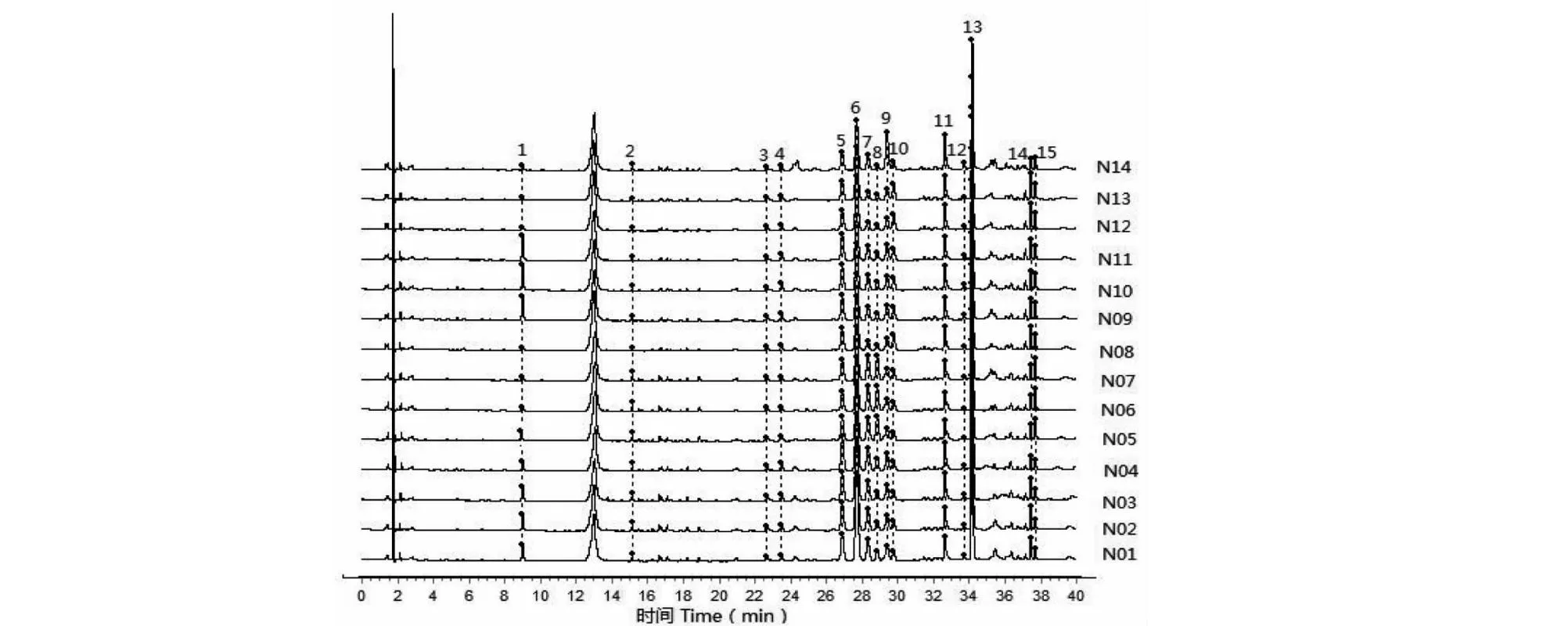
图1 14批南五味子药材UPLC指纹图谱
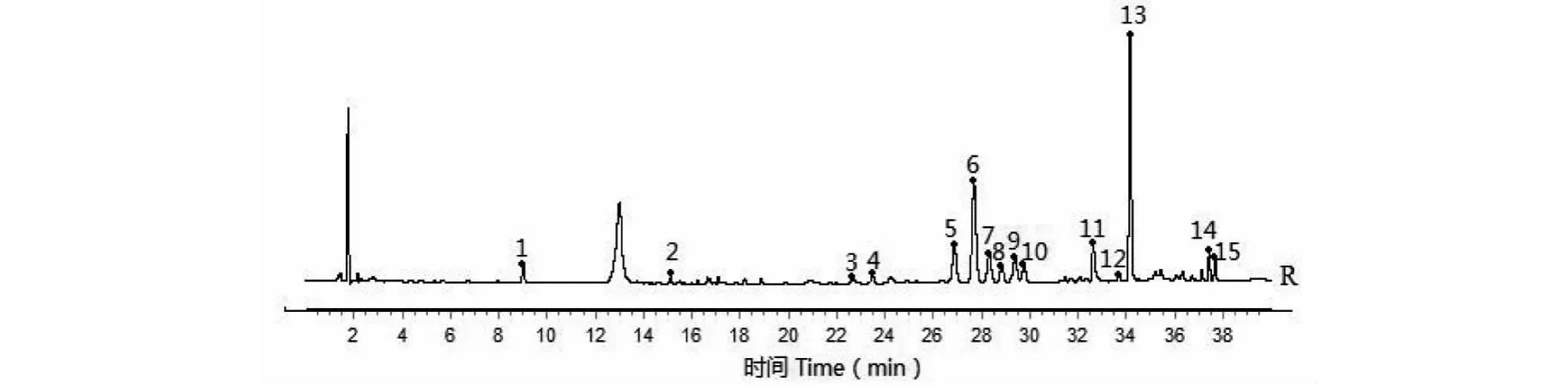
图2 南五味子药材对照指纹图谱

图3 共有指纹峰的指认
2.1.6 相似度评价
采用《中药色谱指纹图谱相似度系统(2012.130723版本)》软件计算14批南五味子药材指纹图谱与对照指纹图谱的相似度(见表2),结果显示,14批南五味子药材指纹图谱的相似度值在0.987~0.996,均在0.95以上,表明指纹图谱可作为共性质量特征,用于南五味子药材的鉴别和质量控制。
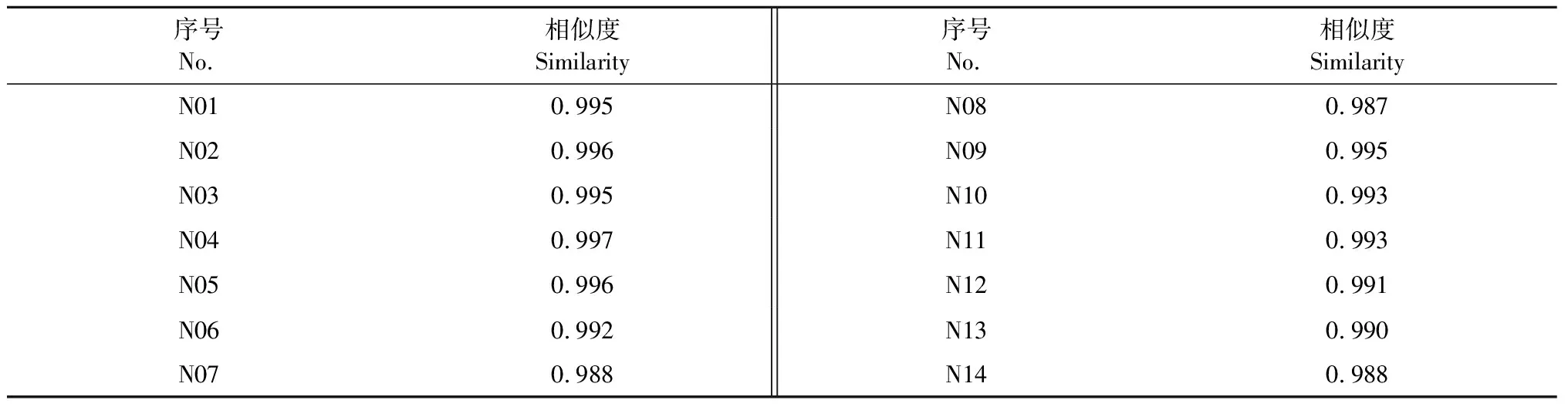
表2 相似度评价结果
2.1.7 化学计量学分析
2.1.7.1 聚类分析(HCA)
采用IBM SPSS Statistics 20.0软件,以14批南五味子药材指纹图谱15个共有峰的峰面积为变量,采用Z得分法对峰面积进行标准化处理,样本间距以平方Euclidean距离表征,以组间联接法对14批南五味子药材指纹图谱进行聚类分析(见图4)。结果显示,组间距离为10~15时,14批南五味子药材样品分为四类,其中编号N01~N05的样品归为第一类;编号N06~N08的样品归为第二类,编号N09~N11的样品归为第三类,编号N12~N14的样品归为第四类,说明不同产地的南五味子药材化学成分的含量存在一定的差异。
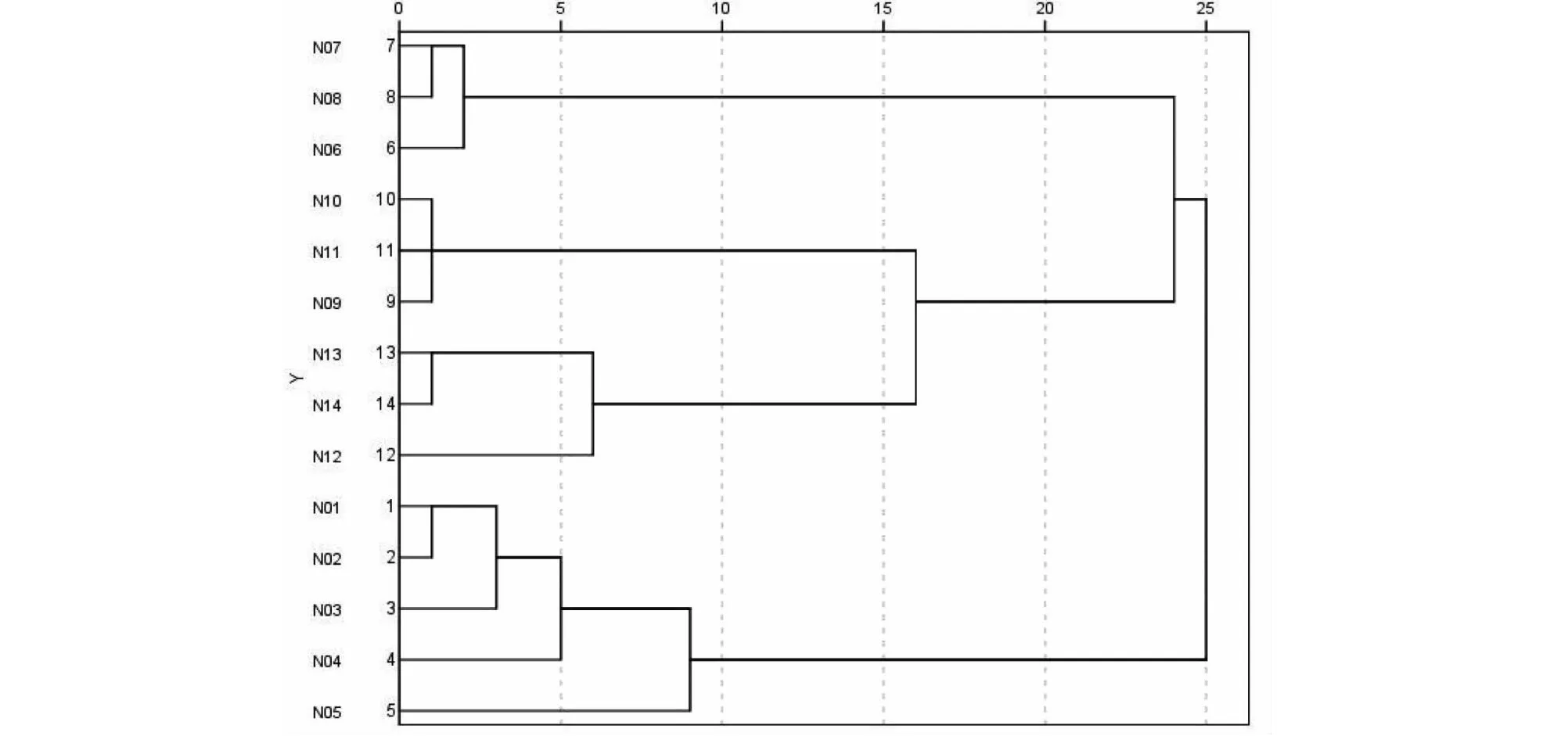
图4 聚类分析图
2.1.7.2 正交偏最小二乘法-判别分析(OPLS-DA)
将14批南五味子样品中各指纹峰的峰面积为变量,采用SIMCA 14.1软件建立OPLS-DA模型,其中R2X=0.882,R2Y=0.953,Q2=0.919,均高于0.5,表明建立的模型稳定有效,可以用于不同产地南五味子的模式识别。通过OPLS-DA的Scores图(见图5)可知,不同产地南五味子药材在模型中空间分布不同,也明显分为4类。且对VIP值(见图6)进行分析可知,共筛选出9个差异标志物,VIP>1.0的依次分别为峰1>峰6>峰12>峰8>峰2>峰4>峰15>峰9>峰7,说明该成分对区分不同产地南五味子药材的贡献较大,其中对峰1(原儿茶酸)对不同产地南五味子样品分类有显著影响。通过对14批南五味子药材峰1(原儿茶酸)的峰面积结果分析,发现河南产地的3批南五味子药材原儿茶酸含量最高,湖北次之,陕西、山西产地最低,这可能与药材产地、加工方式等差异有关。
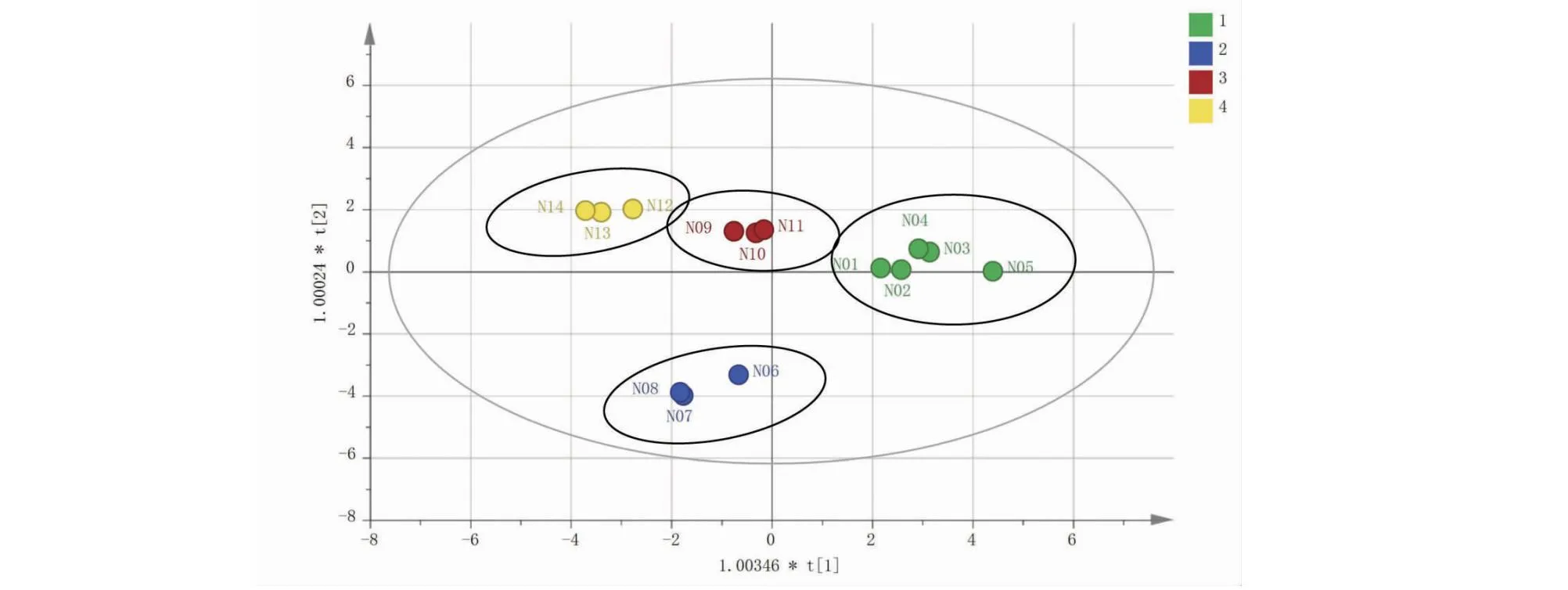
图5 南五味子药材的OPLS-DA分析Scores图

图6 南五味子药材VIP图
2.2 一测多评含量测定的建立
2.2.1 精密度试验
精密吸取“2.1.2”项下混合对照品溶液,按照“2.1.1”项下色谱条件重复进样6次,记录五味子酯丙、五味子酯甲、五味子酯乙、安五脂素、五味子甲素和五味子酯丁6种木脂素类成分的峰面积,并计算其RSD值均小于3.0%,表明仪器精密度良好。
2.2.2 线性关系考察
精密称定五味子酯丙对照品6.586 mg、五味子酯甲对照品6.301 mg、五味子酯乙对照品7.332 mg、安五脂素对照品5.569 mg、五味子甲素对照品8.745 mg和五味子酯丁对照品7.146 mg,加甲醇制成每1mL含五味子酯丙258.171 μg、五味子酯甲239.942 μg、五味子酯乙287.414 μg、安五脂素218.305 μg、五味子甲素348.051 μg、五味子酯丁280.123 μg的混合溶液,即得对照品贮备液。精密移取上述对照品贮备液1、1、1、2、3 mL,分别置100、25、5、5、5 mL量瓶中,加甲醇制成系列浓度的混合对照品应用液;分别取上述混合对照品贮备液和混合对照品应用液,分别按“2.1.1”项下色谱条件依次进样分析,记录色谱峰面积,以峰面积为纵坐标(Y),对照品浓度为横坐标(X),绘制标准曲线,结果见表3,结果显示,各指标成分在相应的浓度内峰面积与对照品浓度线性关系良好。
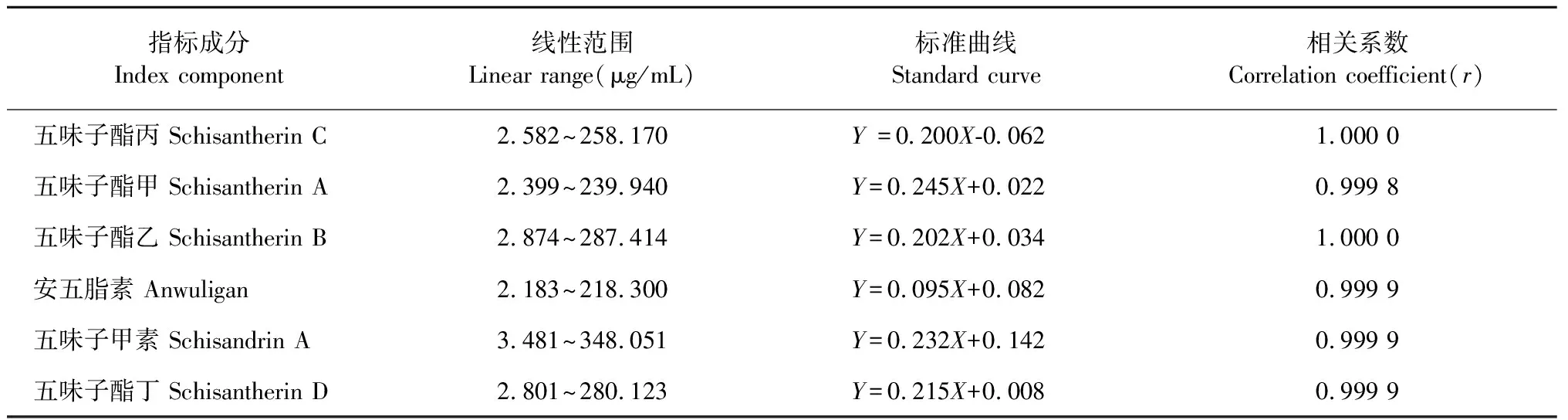
表3 6种成分的线性回归方程及线性范围
2.2.3 重复性试验
取同一份样品(编号:N01,过三号筛)约0.5 g,共6份,精密称定,按“2.1.3”项下方法制备供试品溶液,按“2.1.1”项下色谱条件进样分析,计算五味子酯丙、五味子酯甲、五味子酯乙、安五脂素、五味子甲素和五味子酯丁6种木脂素类成分的峰面积,并计算其RSD值均小于3.0%,表明该方法重复性良好。
2.2.4 稳定性试验
取南五味子药材供试品溶液,按“2.1.1”项下色谱条件,分别在0、2、5、7、12、18、24 h测定,计算五味子酯丙、五味子酯甲、五味子酯乙、安五脂素、五味子甲素和五味子酯丁6种木脂素类成分的峰面积,并计算其RSD值均小于3.0%,表明供试品溶液在24 h内稳定性良好。
2.2.5 加样回收率试验
取已知含量测定的南五味子药材粉末(编号:N01,过三号筛)约0.25 g,精密称定,按样品与对照品含量比为1∶0.5、1∶1及1∶1.5的比例加入五味子酯丙、五味子酯甲、五味子酯乙、安五脂素、五味子甲素和五味子酯丁6种木脂素类成分对照品,每种比例平行3份,按“2.1.3”项下方法制备9份供试品溶液,按“2.1.1”项下色谱条件进样分析,计算6种成分的回收率及RSD值,结果见表4,五味子酯丙、五味子酯甲、五味子酯乙、安五脂素、五味子甲素和五味子酯丁6种木脂素类成分的加样回收率范围分别是99.27%~102.0%、97.20%~101.4%、98.41%~101.8%、97.20%~99.77%、98.74%~101.3%、98.36%~101.2%,RSD值均小于2.0%,表明该分析方法准确度良好。
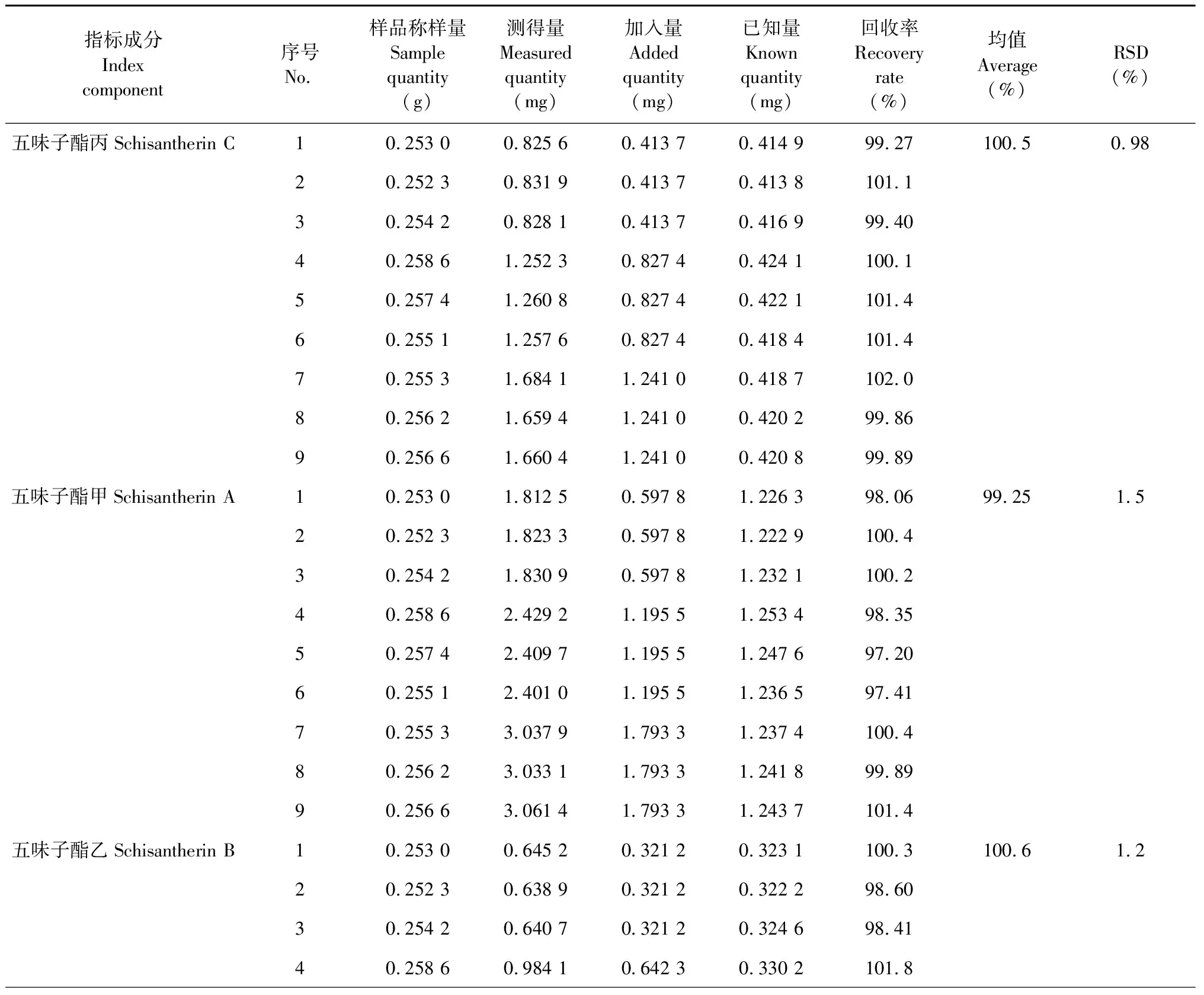
表4 6种成分的加样回收率结果
2.2.6 相对校正因子计算
以五味子酯甲为内标物,根据“2.2.2”项下线性考察结果,以对照品的浓度及其对应的色谱峰面积,分别计算内标物五味子酯甲与五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁的相对校正因子,计算公式为fsi=fs/fi=Cs×Ai/(Ci×As),其中As为内标物s的对照品峰面积,Cs为内标物s的对照品浓度,Ai为其他成分i的对照品峰面积,Ci为其他成分i的对照品浓度。结果五味子酯丙的平均校正因子为1.22,五味子酯乙的平均校正因子为1.21,安五脂素的平均校正因子为2.57,五味子甲素的平均校正因子为1.05,五味子酯丁的平均校正因子为1.14,结果见表5。
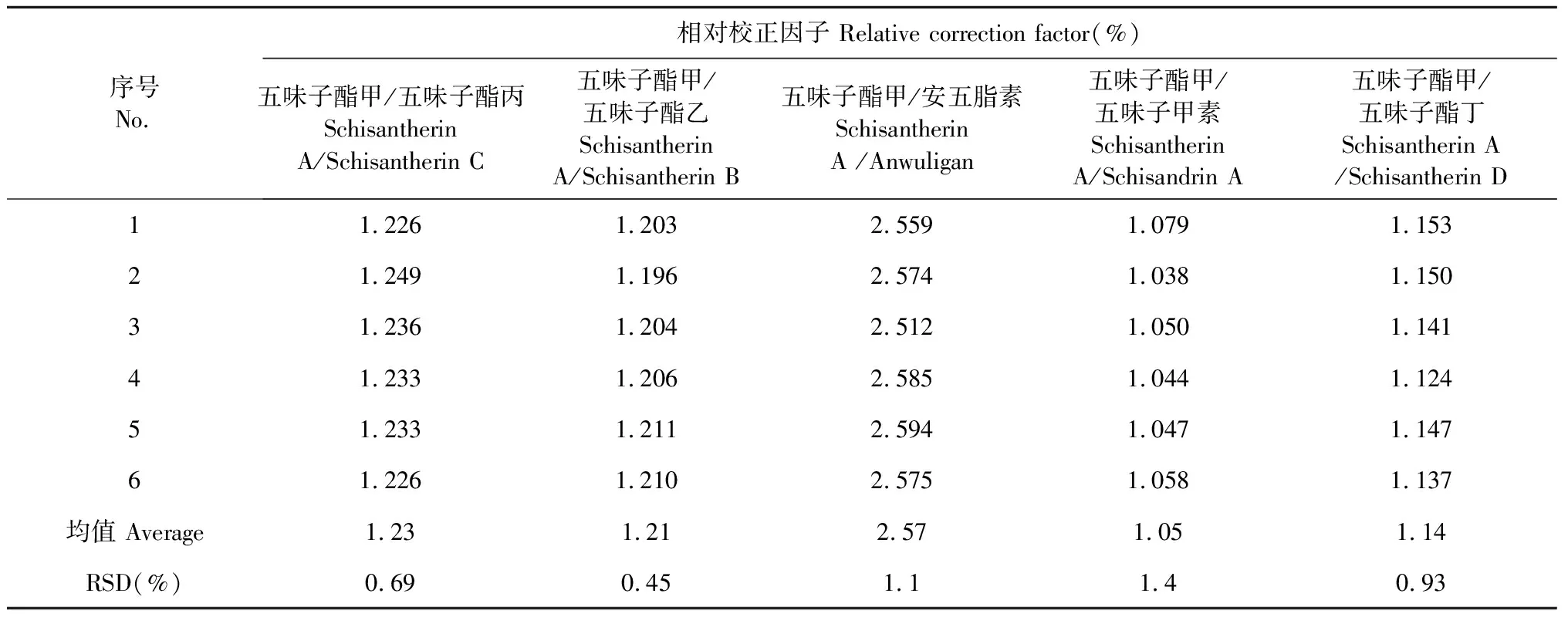
表5 5种成分相对校正因子计算结果
2.2.7 系统适用性试验
2.2.7.1 不同仪器考察
以五味子酯甲为参照,计算五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁在不同品牌仪器上(Waters H-Class、Thermo Vanquish和Agilent 1290)的相对保留时间和相对校正因子,结果见表6。结果显示,上述5种指标成分的相对保留时间RSD在0.00%~0.35%范围内,相对校正因子RSD在0.25%~0.57%范围内,表明不同仪器对五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁的相对保留时间和相对校正因子无显著影响。

表6 不同仪器下相对保留时间和相对校正因子测定结果
2.2.7.2 不同流速考察
以五味子酯甲为参照,计算五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁在不同流速下(0.28、0.30、0.32 mL/min)的相对保留时间和相对校正因子,结果见表7。结果显示,上述5种指标成分的相对保留时间RSD在0.06%~1.2%范围内,相对校正因子RSD在0.41%~2.0%范围内,表明不同流速对五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁的相对保留时间和相对校正因子无显著影响。

表7 不同流速下相对保留时间和相对校正因子测定结果
2.2.7.3 不同柱温考察
以五味子酯甲为参照,计算五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁在不同柱温下(28、30、32 ℃)的相对保留时间和相对校正因子,结果见表8。结果显示,上述5种指标成分的相对保留时间RSD在0.06%~0.23%范围内,相对校正因子RSD在0.47%~1.7%范围内,表明不同柱温对五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁的相对保留时间和相对校正因子无显著影响。
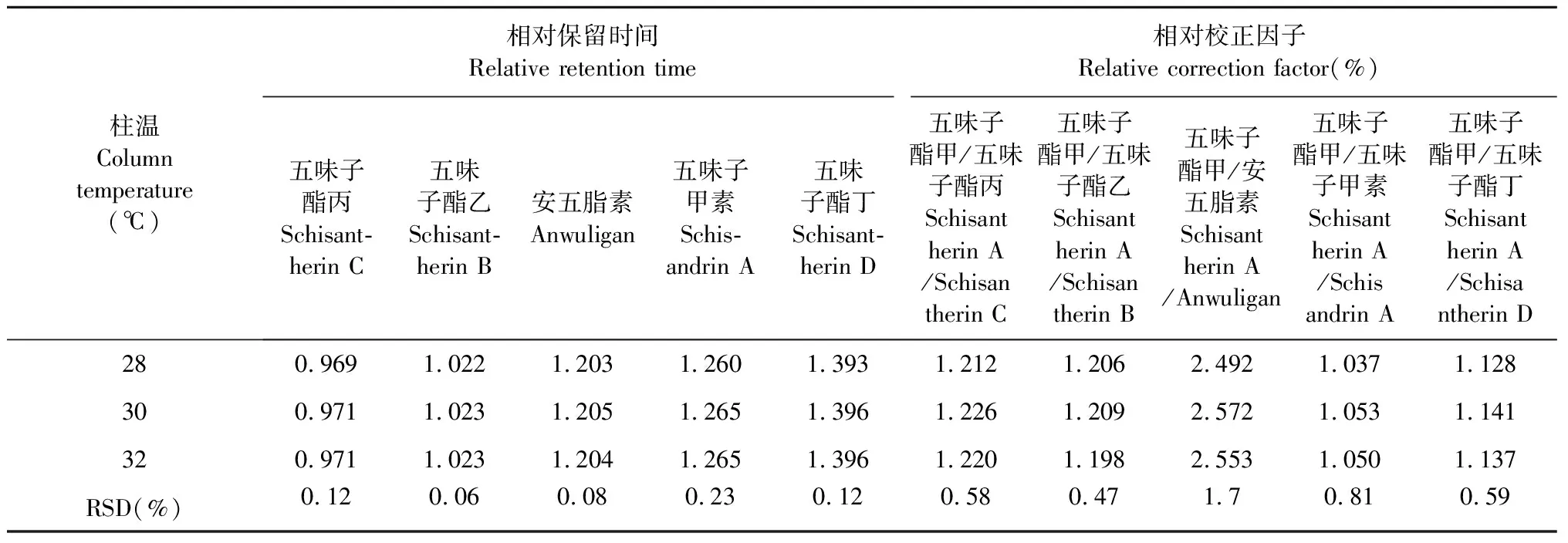
表8 不同柱温下相对保留时间和相对校正因子测定结果
2.2.8 色谱峰定位
取“2.2.7系统适用性”项下不同仪器、不同流速和不同柱温的相对保留时间均值,确定五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁的相对保留时间分别为0.97、1.0、1.2、1.3和1.40。
2.2.9 样品测定
取14批南五味子药材,按“2.1.3”项下供试品溶液制备方法和“2.1.1”项下色谱条件对14批南五味子进行测定,以五味子酯甲为参照,分别采用外标法(EMS)和一测多评法(QAMS)计算五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁的含量,并比较两种计算方式的相对偏差,结果见表9。结果显示,两种方法测定的五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁的含量相对偏差均小于3.0%,说明两种方法计算结果无显著差异,QAMS准确度良好。根据外标法计算结果可知,湖北产地的5批南五味子药材6种木脂素总含量最高,其余9批陕西、河南、山西产地的样品含量较均匀,其中山西产地3批含量最低。
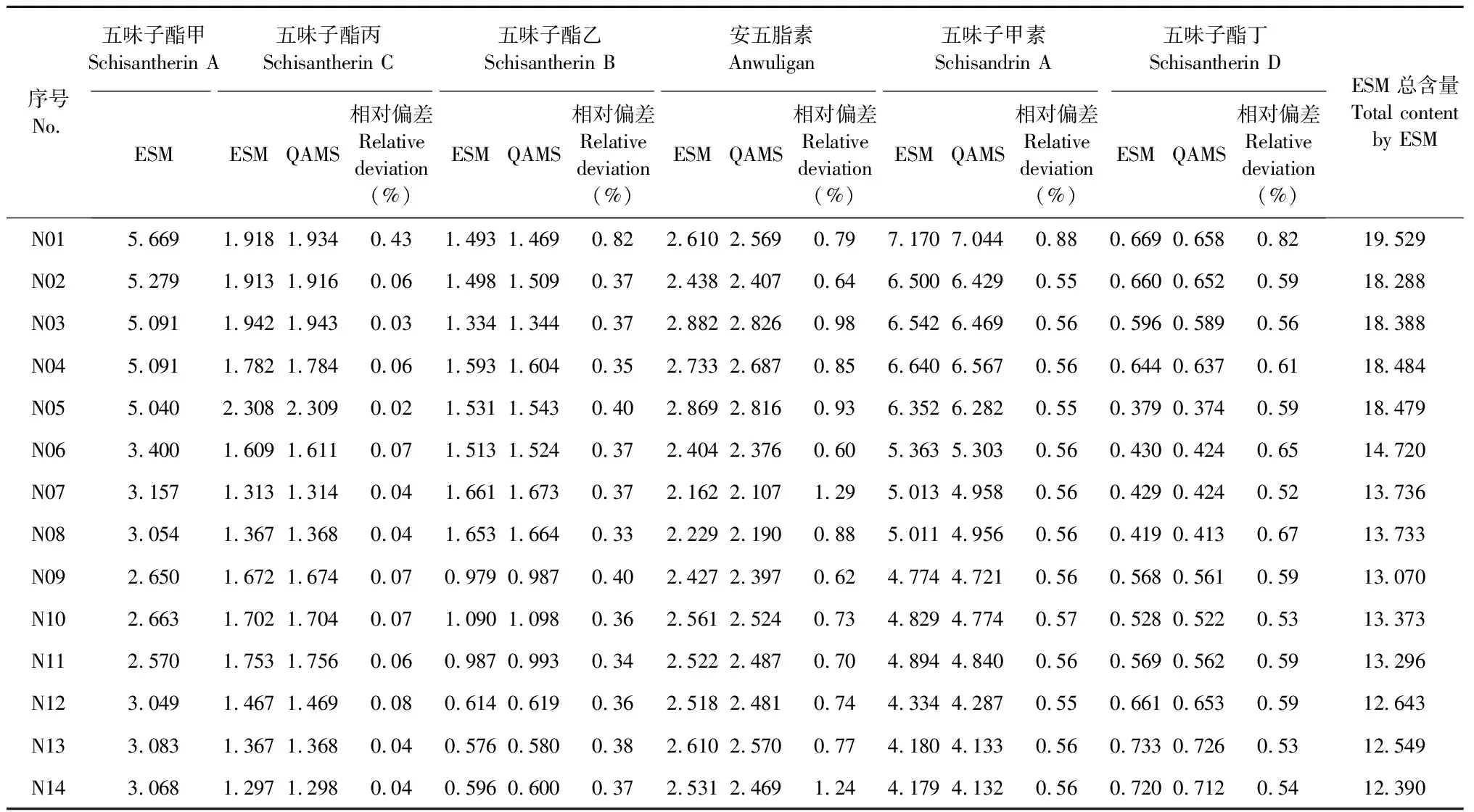
表9 14批南五味子药材含量测定结果
3 讨论与结论
2020年版《中国药典》规定南五味子药材在秋季果实成熟时采摘。有文献报道,Li等[10]对湖北、四川、江西三个产区南五味子药材中主要化学成分的含量变化进行研究,通过对测定结果分析,发现三个产区不同生长期的南五味子药材中主要化学成分积累具有一定的规律;初步确定南五味子的最佳采收期为9月中下旬。Huang等[11]采用紫外分光光度法和液相色谱法,测定不同采收期陕西产南五味子中挥发油、总木脂素、五味子酯甲和五味子甲素的含量,结果认为南五味子最佳采收期在8月上旬为佳。此外,研究显示,不同的产地加工方式对南五味子有效成分的含量有显著影响[12]。本次研究发现,湖北产地的5批南五味子药材6种木脂素总含量最高,其余9批陕西、河南、山西产地的样品含量较均匀,其中山西产地3批含量最低;此外,原儿茶酸含量最高的则是河南产地,湖北次之,陕西、山西产地最低,这可能与药材的产地、采收时间、加工方式等差异有关。
本研究采用UPLC建立南五味子药材指纹图谱,分别考察了不同流动相系统(甲醇-水、乙腈-水、乙腈-0.1%磷酸),结果显示,采用乙腈-0.1%磷酸为洗脱系统,色谱峰的信息较全面,分离效果较好。采用二极管阵列检测器(DAD检测器)对南五味子药材供试品溶液在190~350 nm范围内进行全波长扫描,并提取不同波长(230、254 nm)下的指纹图谱进行对比,结果显示,在230nm下,指纹图谱色谱峰的响应和色谱峰容量均较好。还考察了不同柱长[Waters ACQUITY HSS T3(2.1 mm×100 mm,1.8 μm)、Waters ACQUITY HSS T3(2.1 mm×150 mm,1.8 μm)]对南五味子药材指纹图谱各色谱柱的分离度,结果显示,Waters ACQUITY HSS T3(2.1 mm×150 mm,1.8 μm)色谱柱分离度较好。此外,通过单因素分析,以“指纹图谱总峰面积/称样量”及6种木脂素类成分的含量为评价指标,分别考察了提取溶剂、提取方式和提取时间对南五味子药材指纹图谱及含量测定的影响,确定最佳供试品溶液制备方法,使指纹图谱各色谱峰提取效率达到最高。
目前,指纹图谱相似度评价法、欧氏距离聚类分析法和主成分分析法等化学模式识别方法在中药材质量、产地差异以及品种鉴别等研究领域的应用已较为广泛[13-15]。本研究引入指纹图谱相似度评价分析、聚类分析以及正交偏最小二乘法-判别分析,可以直观衡量不同批次南五味子样品间指纹图谱变化模式的相似性及亲疏程度。结果显示14批南五味子药材指纹图谱与对照指纹图谱相似度均在0.95以上,说明不同产地南五味子药材有较好的一致性。通过聚类分析和正交偏最小二乘法判别分析,可将14批南五味子药材分为4类,呈现一定的产地规律性;共筛选出9个差异性物质,分别为峰1(原儿茶酸)、峰2、峰4、峰6、峰7(五味子酯乙)、峰8、峰9、峰12、峰15,说明以上9个成分对南五味子指纹图谱影响较大。有待进一步深入研究。本实验利用南五味子木脂素类化合物结构的相似性,建立了采用QAMS法对南五味子药材中的6种木脂素类成分的含量进行初步测定。此次研究选择的五味子酯甲、五味子酯丙、五味子酯乙、安五脂素、五味子甲素和五味子酯丁成分在南五味子药材具有一定的生物活性,可以反映药材的内在质量。其中五味子酯甲化学性质稳定,作为内参物,具有一定的代表性。通过比较一测多评法(QAMS)和外标法的相对误差,结果表明所建立的南五味子“一测多评”含量测定方法与外标法的测定结果无显著性差异,表明建立的方法具有良好的可信度。
本研究采用指纹图谱与一测多评相结合的手段,从定性、定量两方面来评价南五味子药材的质量,可为提升南五味子药材质量标准提供参考。
猜你喜欢
中国粮油学报(2019年4期)2019-07-12
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年4期)2018-05-07
中成药(2017年3期)2017-05-17
中成药(2017年3期)2017-05-17
中成药(2016年8期)2016-05-17
中国继续医学教育(2015年2期)2016-01-06
药学研究(2015年11期)2015-12-19
中国药理学通报(2014年2期)2014-05-09
食品工业科技(2014年7期)2014-03-11
