
伴锥体束损害及眼震快速进展型重症肌无力一例临床分析
2019-10-09 11:45翁映虹笪宇威徐敏卢岩徐佳丽邱占东贾宇赵红梅
中国全科医学 2019年30期
翁映虹,笪宇威,徐敏,卢岩,徐佳丽,邱占东,贾宇,赵红梅
重症肌无力(MG)是由以乙酰胆碱受体抗体(AChR-Ab)为主的多种抗体介导,以体液免疫为主,细胞免疫为辅,补体参与,涉及整个免疫网络功能异常的一种获得性自身免疫性疾病[1-3]。MG可发生于任何年龄,影响着全球约70多万人[4],其年发病率为(0.3~2.8)/10万[5]。该病为可治性疾病,如能早诊断并及时根据患者情况予个体化治疗,可避免进一步加重出现肌无力危象危及生命,故正确认识和早期诊断MG对改善患者预后极为重要。但临床上MG不典型的表现较多,较难鉴别。本文通过分析1例伴锥体束损害及眼震的快速进展型MG的临床及预后,并对相关文献进行复习,总结该类患者临床特点、诊断、治疗及预后情况,旨在提高临床医师对本病的认识。
1 资料与方法
1.1 一般资料 患者为42岁女性,因“双眼睑下垂伴视物成双4 d,双下肢无力伴言语含糊1 d” 于2018-06-28至首都医科大学宣武医院就诊,收入神经内科。患者4 d前无明显诱因突发双眼睑下垂,视物成双,呈持续性,休息无缓解,至外院查颅脑CT、MR检查显示无异常,未行进一步诊治。2 d前患者上述症状加重并呈晨轻暮重,1 d前出现说话含糊,轻度吞咽困难,双下肢无力,蹲起困难,晨轻暮重。自觉双上肢有力,无肢体麻木、疼痛、萎缩。既往史:10年前曾一过性视物成双,数天后自行好转,未系统诊治。
1.2 研究方法 分析本例伴锥体束损害及眼震的快速进展型MG患者的临床资料,结合文献回顾分析伴锥体束损害、眼震快速进展型MG的临床特点、诊断、治疗及预后,并探讨其发病机制。
2 结果
2.1 临床特点
2.1.1 入院神经系统查体 患者主诉“双眼睑下垂伴视物成双4 d,双下肢无力伴言语含糊1 d”,入院查体为构音欠清,双睑下垂,眼裂位于10:00~14:00水平,视力正常,双眼右视粗大水平眼震,左视细小水平眼震,下视旋转眼震,闭目力弱,鼓腮无力,咽反射存在,舌肌无力,面部浅感觉减退,颈伸、屈肌力4级,余脑神经正常。四肢近端3~4级(双下肢重),远端5级。四肢肌张力正常、腱反射亢进,双手霍夫曼征阳性,右掌颌反射阳性,左掌颌反射阴性,双侧胸大肌反射、腹直肌反射阳性。左踝阵挛阳性,右踝阵挛阴性,左下肢巴氏征可疑阳性,右下肢巴氏征阳性。无肢体感觉障碍。脑膜刺激征阴性。新斯的明试验(0.02 mg/kg):阳性,30 min双上睑平视相对计分改善60%,双下肢疲劳试验相对计分改善50%。MG定量评分体系(QMG)评分16分,MG日常生活能力量表(MG-ADL)评分10分,MG综合评分17分。
2.1.2 实验室检查 血、尿常规,生化全项、糖化血红蛋白、风湿三项、自身免疫相关抗体谱、肿瘤标志物正常,北京协和医院MG抗体(放射免疫沉淀法):AChR-Ab、Musk-Ab均阴性。血液及脑脊液(CSF)神经节苷脂抗体谱阴性。丙种球蛋白(IVIg)治疗前及冲击完成后第4天查CSF压力110~120 mm H2O(1 mm H2O=0.098 kPa),CSF常规、生化、TORCH10项、自身免疫性脑炎、CSF IgM、CSF特异IgG寡克隆区带(OB)、24 h CSF IgG鞘内合成率均在参考范围内,血及CSF副肿瘤标志物在参考范围内。涂片均未见细菌、隐球菌、抗酸杆菌。治疗前CSF及血IgG OB弱阳性,IVIg冲击完成后为阴性。
2.1.3 电生理及影像检查 电生理检查:双正中、尺、胫、腓总神经运动、感觉传导未见异常,右胫神经、右正中神经F波未见异常,右胫神经H反射未见异常。治疗前重复频率电刺激(RNS):右面神经低频5 Hz下降16%,右副神经低频3 Hz下降21%、5 Hz下降23.6%。IVIg冲击完成后第5天复查RNS:右面神经3 Hz下降46.3%,5 Hz下降55%;右副神经3 Hz下降27.3%、5 Hz下降31.3%,低频递减较前明显(见图1)。治疗前及IVIg冲击完成后第4天查颅脑MR平扫+增强检查未见异常。胸部CT平扫+增强检查:胸腺退化不全?2.2 诊断、治疗及预后 根据患者急性起病,眼外肌、球部、面肌、颈肌、四肢肌无力,呈晨轻暮重,疲劳试验阳性,新斯的明试验阳性,RNS低频递减,停用溴吡斯的明后症状明显加重,恢复溴吡斯的明治疗后症状减轻,诊断为MG〔Osserman分型ⅡB,美国重症肌无力协会(MGFA)分型ⅢA〕。根据患者病情及体质量,予个体化治疗。予溴吡斯的明 120 mg/次,4次 /d、IVIg 0.4 g·kg-1·d-1×5 d治疗,IVIg冲击完成后予醋酸泼尼松片20 mg逐渐递增至40 mg,病情平稳后加予甲氨蝶呤并逐渐减量溴吡斯的明至停用。IVIg冲击完成后第2天患者双下肢巴氏征消失,出院时QMG评分9分,MG-ADL评分8分,MG综合评分9分。3个月后患者症状及阳性体征全部消失,之后随访2个月未复发。
3 讨论
3.1 临床特点、治疗及预后 本例患者急性起病,进展快速,除了眼外肌、球部、四肢肌无力,晨轻暮重,易疲劳等典型的MG临床表现外,还有面肌无力,眼震,面部浅感觉减退,腱反射亢进,病理征阳性。新斯的明试验阳性、RNS低频递减10%以上,AChR-Ab阳性,胸腺退化不全,无其他CNS病变依据。经胆碱酯酶抑制剂、激素、IVIg、免疫抑制剂治疗后症状快速好转,之后症状完全消失,随访无复发,预后良好。
3.2 鉴别诊断 本病例诊断MG明确,但存在锥体束损害、眼震、面部痛温觉减退,CSF OB弱阳性,对诊断造成一定干扰。需与以下疾病相鉴别。(1)Bickerstaff脑干脑炎:为脑干炎性反应,临床表现为急性或亚急性起病,对称性眼肌麻痹、共济失调、意识障碍、腱反射活跃、病理征阳性。血GQ1b抗体阳性,MR检查示脑干异常信号。本例急性起病,眼震、腱反射活跃、病理征阳性。但该患者无前驱感染、共济失调、意识障碍。CSF常规、生化正常,血GQ1b抗体阴性,多次颅脑MR检查正常,故不支持该诊断。(2)Miller-fisher综合征:为吉兰-巴雷综合征变异型,急性起病,数天或数周达高峰,有自限性,可累及多个脑神经,以眼肌麻痹、共济失调,腱反射消失,肢体肌力正常或减退为主要表现。CSF出现蛋白-细胞分离,大多数血GQ1b抗体阳性,电生理有感觉传导速度的波幅和速度下降。本例急性起病,对称累及多脑神经支配区,四肢近端无力。但无CSF蛋白-细胞分离,感觉神经传导正常,血GQ1b抗体阴性,故不支持该诊断。(3)多发性硬化(MS):MS最常见的眼球运动障碍是眼震和核间性眼肌麻痹,可为首发症状。MS累及中枢神经系统(CNS)白质,有时间、空间多发性,临床表现多样,有自限性。CSF中OB阳性,影像学检查可提供亚临床病灶的客观证据。其中临床孤立综合征为单次发病。本例患者急性起病,单次发病,四肢无力、复视及眼震,腱反射亢进、病理征阳性,面部痛温觉减退,持续>24 h后可恢复,CSF中OB弱阳性。但多次颅脑MR检查未见异常,血OB弱阳性而非缺失,故不支持该诊断。
3.3 文献分析 查阅文献,近年研究发现MG除累及骨骼肌神经肌肉接头外,还可累及CNS[6-7]、周围神经系统[8]及自主神经系统[9-10]。
但目前尚无MG同时伴锥体束损害及眼震的相关报道。SHARMA等[11]报道了1例MG伴锥体束损害合并甲状腺功能亢进的患者,病程6个月,眼外肌、面肌、四肢肌、膀胱肌无力,易疲劳,四肢腱反射亢进,腹壁反射消失,双下肢巴氏征阳性。胸腺增生、新斯的明试验、电生理支持MG诊断。经卡比马唑及新斯的明治疗4 d后腱反射恢复正常,双下肢巴氏征消失。张华等[12]报道644例MG患者中有11例伴锥体束损害,且随MG好转而消失。欧阳宛炯[13]报道1例MG伴锥体束损害的患者,病程11 d,左眼睑下垂,张口、吞咽困难,左上肢、双下肢无力,晨轻暮重,伴面部麻木,双下肢巴氏征阳性;RNS低频递减,新斯的明试验阳性,且肌肉注射新斯的明30 min双下肢巴氏征消失;诊断MG,予IVIg冲击5 d,溴吡斯的明10 d后,除左眼睑下垂外其余症状均消失。庞英等[14]报道了1例急性起病的MG伴锥体束损害的患者,病程2 d,眼外肌、球部、面肌、四肢肌无力,双下肢腱反射活跃,左下肢巴氏征阳性,AChR-Ab阳性、RNS低频递减支持MG诊断。该患者无晨轻暮重、CSF OB阳性、新斯的明试验阴性,易干扰诊断。经IVIg、吡啶斯的明治疗16 d后病理征消失。本例患者与上述报道均有眼外肌、球部、面肌、颈肌、四肢肌无力,晨轻暮重,伴锥体束损害,面部浅感觉减退,CSF OB阳性[12,14-15],无其他CNS病变证据,经胆碱酯酶抑制剂、激素、IVIg及免疫抑制剂治疗后可消失,预后较好。但本例患者为急性起病快速进展且同时伴有眼震,病情更重,报道多为亚急性或慢性病程,仅1例患者为急性起病,且均无眼震。
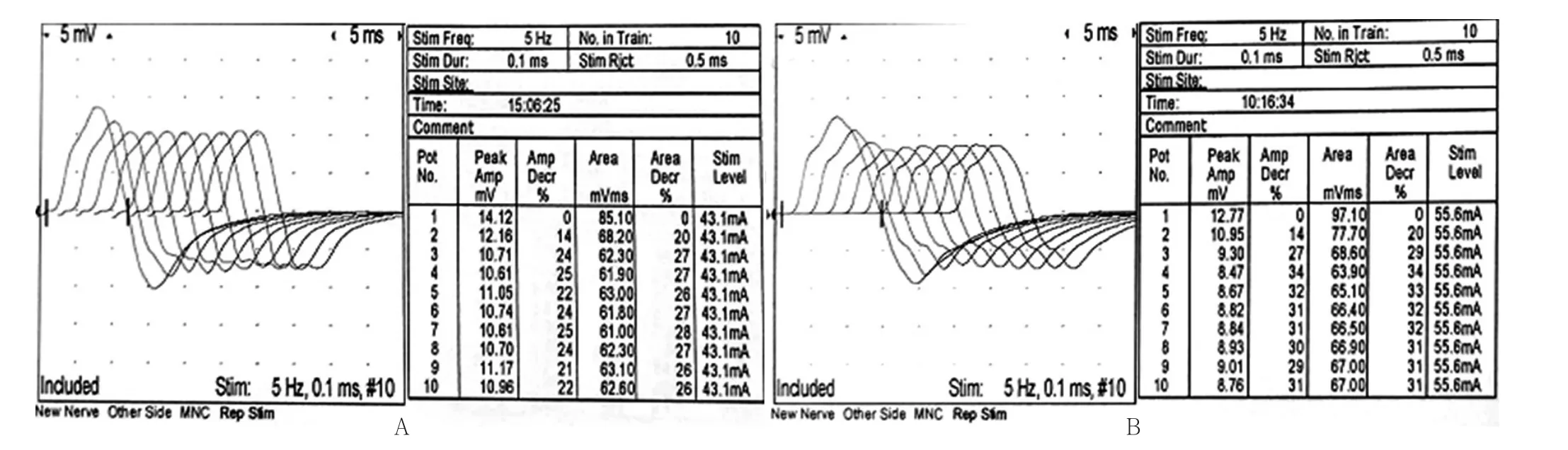
图1 IVIg治疗前后副神经重复频率电刺激结果Figure 1 Results of accessory nerve repetitive nerve stimulation test before and after intravenous immunoglobulin pulse therapy
MG伴眼震的报道因其症状与核间性眼肌麻痹相似,但为内侧纵束以外的病变引起而在国外多称为MG合并假性核间性眼肌麻痹[16-19]。NIJSSE等[16]报道1例波动性复视、左眼睑下垂患者,有核间性眼肌麻痹相似症状,但无CNS损害,AChR-Ab阳性,诊断眼肌型MG,经新斯的明治疗后症状消失。ACERS[17]报道1例眼肌型MG伴水平眼震的患者,病程1个多月,波动性复视,新斯的明试验阳性,予新斯的明治疗后症状完全消失。ITO等[18]报道1例MG合并双侧假性核间性眼肌麻痹的患者,有波动性复视、耳鸣、平衡失调,之后出现波动性吞咽困难、双上肢无力,易疲劳,有类似核间性眼肌麻痹的眼震,但外展时旋转性眼震,AChR-Ab阳性,诊断为MG,胆碱酯酶抑制剂可缓解症状,经胸腺切除后眼球运动恢复正常。KHANNA等[19]研究了2例眼肌型MG伴水平性眼震患者,并与MS及健康人的眼震相比较,认为MG水平眼震峰速度与健康人相似或稍快,MS内收时眼震速度最慢。国内仅1篇相关报道,林书乐等[20]报道了2例眼肌型MG伴水平性眼震的患者,均为慢性病程,经胆碱酯酶抑制剂治疗后眼震及其他阳性体征均消失,预后较好。报道中的患者均无其他CNS病变,经胆碱酯酶抑制剂、胸腺瘤切除后症状可消失,本文与其一致。但报道多数为眼肌型MG合并水平眼震,较少全身型MG、旋转性眼震的报道,病程多进展缓慢,而本例患者为全身型MG伴水平、旋转性眼震,迅速进展至球部,病情较重,要求临床医生能快速识别、正确诊断并迅速拟定精准的个体化治疗方案。
3.4 发病机制 MG伴可逆性锥体束损害的机制尚不明确。主要有两个观点:(1)认为MG是一种受体病,不仅局限于神经肌肉接头,还可累及其他系统。早期有学者通过MG患者CSF与血清中IgG与AChR-Ab比值证实AChR-Ab来源于鞘内[21]。认为可能由于AChR-Ab与中枢神经烟碱型AChR(n-AChR)结合,阻碍Ach与n-AChR结合而产生一系列免疫反应[22]。李柱一等[23]通过建立动物MG的CNS受损模型研究AChR-Ab与n-AChR之间的免疫反应,发现MG抗肌-nAChR-Ab不仅可与外周AChR结合引起肌无力等症状,还可与n-AChR结合直接作用于CNS,引起CNS功能障碍,如AChR-Ab作用于CNS锥体束途径中n-AChR而导致锥体束征。(2)认为可能是自身免疫应答泛化所致。张华等[12]发现此类患者CSF鞘内IgG浓度及合成率均高于不伴锥体束损害的MG患者及健康人群。推测鞘内合成的IgG中有与锥体束通路相关的免疫应答导致锥体束损害。
MG伴眼震的发病机制尚不明确。KHANNA等[19]认为其发病机制可能为MG在眼球运动过程中神经肌肉传递冲动失败而发生肌肉疲劳,或神经肌肉传导冲动过程中选择性的保留了眼外肌纤维。有学者认为这是一种中枢代偿作用,克服内收力弱的一个符合Hering's神经支配定律的适应过程[16,18]。
本例患者MG伴可逆性锥体束损害及眼震的发病机制可能为AChR-Ab与n-AChR结合产生免疫反应出现椎体束损害,影响眼球运动中神经肌肉接头传递,出现眼肌疲劳,从而产生眼震。当免疫治疗后,AChR-Ab与n-AChR正常结合,神经肌肉接头传递恢复,则椎体束损害及眼震消失。
4 总结
综上所述,MG伴锥体束损害及眼震的临床特点除了典型的MG常见眼外肌、球部、四肢肌无力,晨轻暮重,易疲劳等表现外,还可有面肌、膀胱肌等无力,水平和(或)旋转性眼震,面部浅感觉减退,腱反射可活跃或亢进,病理征阳性,新斯的明试验阳性、RNS低频递减10%以上和(或)AChR-Ab阳性,可有胸腺增生或胸腺瘤,无其他CNS病变,可为急性、亚急性或慢性病程,经胆碱酯酶抑制剂、激素、IVIg、免疫抑制剂或胸腺切除术等治疗后症状可好转或消失,预后良好。临床上需注意与脑干脑炎、Miller-fisher综合征、MS等鉴别。
随着对MG的不断探索,本研究团队认为:(1)MG可能是一种AChR-Ab相关的全神经系统均可受累的自身免疫性疾病,而非单一神经肌肉接头受累。(2)目前MG临床上常用的Osserman分型及MGFA分型[24]均未将神经肌肉接头受累以外的其他临床表现纳入分型中,随着MG临床观察资料的积累,可期在诊治指南的MG分型中增加合并其他神经系统受累的分型,以便临床医生更好地掌握MG的临床表现形式,避免误诊漏诊,指导治疗。
猜你喜欢
听力学及言语疾病杂志(2022年1期)2022-11-27
中国耳鼻咽喉颅底外科杂志(2022年3期)2022-11-10
黑龙江医药科学(2021年4期)2021-11-16
中华养生保健(2021年18期)2021-02-13
世界科学技术-中医药现代化(2020年2期)2020-07-25
影像研究与医学应用(2020年5期)2020-04-02
中国现代神经疾病杂志(2020年1期)2020-01-08
中国中医急症(2019年10期)2019-05-21
现代养生·下半月(2016年4期)2016-10-21
安徽医药(2015年1期)2015-03-22
