
FAD依赖的葡萄糖脱氢酶多拷贝毕赤酵母菌株的构建及高效表达
2023-12-09 03:29罗同阳高庆华王庆庆
微生物学杂志 2023年4期
董 聪, 王 玥, 罗同阳, 高庆华, 王庆庆
(河北省微生物研究所有限公司,河北 保定 071051)
FAD依赖的葡萄糖脱氢酶(FAD-GDH)(EC1.1.99.10)是一类以FAD为辅基催化D-葡萄糖氧化生成葡萄糖酸-δ-内酯的氧化还原酶,广泛应用于血糖检测、新型燃料电池[1]、植入式心脏起搏器[2]等领域,特别是在血糖检测方面,由于FAD-GDH不利用氧作为电子受体,故不受血液氧分压影响[3],适用于检测静脉血、动脉血和高海拔等氧含量不同的样本[4]。基于FAD-GDH的血糖计不依赖于氧,对红细胞压积的干扰小,对麦芽糖、木糖等不敏感,避免了其他糖类干扰产生错误结果影响治疗,近年来成为便携式血糖监测系统的研究热点。毕赤酵母(Pichiapastoris)表达系统是目前使用最多和最广泛的外源蛋白表达的工具之一[5-6],FAD-GDH在毕赤酵母表达系统中有过实现可溶表达的报道[7]。周利伟[8]、杨愈丰[9]、Yang等[10-11]、郭坚等[12]将FAD-GDH在毕赤酵母中成功表达且进行了结构、酶学性质等一系列研究。但是,上述研究在构建重组表达菌株时转入的都是含单拷贝基因的表达载体,转化毕赤酵母后表达FAD-GDH能力有限。诸多研究表明提高蛋白表达的方法有很多,提高基因拷贝数尤为重要[13-14]。Cámara等[15]为提高成熟肽mROL的表达量,利用同尾酶法成功构建最高含15个目的基因拷贝数的毕赤酵母重组菌株。Teng等[16]为提高腐生子囊菌(Pseudoplectanianigrella)防御素(plectasin)的表达量,构建了包含8拷贝表达盒的重组表达载体,其在毕赤酵母中的表达量是单拷贝表达量的1.63倍。吕星星等[17]构建了鳜鱼β-防御素2拷贝、4拷贝重组表达菌株,在毕赤酵母中提高了表达量。王义春等[18]构建含1~6个玉米赤霉烯酮降解基因表达盒的表达质粒,获得ZEN降解酶重组菌株,4拷贝的转化子表达水平最高,三角瓶发酵3 d,酶活达到10 U/mL。近年来,我们通过密码子优化等方法构建了含单个表达盒的产FAD依赖的葡萄糖脱氢酶的重组毕赤酵母X33/pMD-GDH,在10 L发酵罐水平经甲醇诱导表达后,酶活为257.6 U/mL,且该酶具有良好的酶学性质[19]。本研究旨在前期研究的基础上,利用同尾酶法构建基因的多拷贝表达盒载体策略,构建基因多拷贝重组表达菌株;同时通过qRT-PCR分析确定各重组菌株基因中目的基因拷贝数[20],以考察研究不同拷贝数对酶活表达的影响,以期实现高效表达。
1 材料与方法
1.1 材料
1.1.1 菌株和载体 大肠埃希菌(Escherichiacoli)TOP10购自天根生化科技有限公司(北京);酵母表达菌株、毕赤酵母(Pichiapastoris)X33由中科院微生物研究所提供;质粒pMD-GDH由河北省微生物研究所有限公司分子生物学实验室(本实验室)前期构建,质粒pPICZαA购自淼灵生物。
1.1.2 培养基及显色液 ①大肠埃希菌LB 培养基、毕赤酵母YPD、YPDS 培养基见Invitrogen 公司毕赤酵母操作手册。②YPCS发酵培养基:胰蛋白胨20 g,酵母提取物10 g,酪蛋白水解物5 g,山梨醇5 g,ddH2O 1 L,120 ℃灭菌20 min。③BMGY培养基:胰蛋白胨20 g/L,酵母提取物10 g/L,磷酸氢二钾3 g/L,磷酸二氢钾11.8 g/L,甘油10 mL,加水至895 mL,120 ℃灭菌20 min,温度下降至60 ℃之后,超净台加入100 mL过滤除菌10×YNB(13.4 g/L),2 mL过滤除菌500×生物素(4×10-4g/L),甲醇5 mL。④BSM基础盐培养基:85%磷酸26.7 mL,硫酸钙0.93 g/L,硫酸钾18.2 g/L,硫酸镁14.9 g/L,氢氧化钾4.13 g/L,甘油40 g/L。⑤MD培养基:葡萄糖2 g,琼脂粉2 g,ddH2O 90 mL,灭菌后加入10×YNB 10 mL和500×生物素0.2 mL。⑥MM培养基:琼脂粉2 g,ddH2O 90 mL,灭菌后加入10×YNB 10 mL、500×生物素0.2 mL和0.5 mL甲醇。平板筛选显色液:溶液Ⅰ:浓度为9×103mol/L的2,6-二氯靛酚钠DCIP;溶液Ⅱ:10%葡萄糖(10 g葡萄糖溶于100 mL去离子水中);将10 mL的1%的琼脂糖溶化后加入2 mL溶液Ⅱ、300 μL溶液Ⅰ即为显色液(现用现配)。
1.1.3 主要试剂与仪器设备 Q5DNA聚合酶、限制性内切酶(EcoRⅠ、NotⅠ、BamHⅠ、BglⅡ、DpnⅠ)、虾碱性磷酸酶rSAP和T4DNA连接酶等购自NEB公司;DNA分子量标准购自康为生物科技公司;qRT-PCR试剂SimpleChIP Uniwersal qPCR Master Mix 购自北京百灵克生物科技有限公司;质粒提取试剂盒购自全式金生物科技公司;酵母DNA提取试剂盒、胶回收试剂盒购自天根生化科技有限公司(北京);博来霉素(Zeocin)购自索莱宝生物科技有限公司;YNB(分子级)购自酷来搏生物科技有限公司;生物素购自博奥拓达生物有限公司;DCIP购自BBL Life Sciences;其余试剂为分析纯。引物合成和测序由北京中科希林生物科技有限公司完成。电泳仪(PP-1150,北京凯元信瑞仪器有限公司);PCR扩增仪(ETC811,北京东胜创新生物科技有限公司);紫外凝胶成像仪(Tanon-52 OOMulti,北京原平皓生物科技有限公司);实时荧光定量PCR仪(Thermofisher QuantStutio 1,美国Thermo Fisher Scientific 公司);电击转化仪(Bio-rad micropulser,美国BIO-RAD公司);紫外分光光度仪(L3S,上海仪电仪器);恒温振荡摇床(ZWY-240,上海智诚公司);10 L发酵罐(TGFB300,上海洋格设备公司)。
1.2 方法
1.2.1 重组表达载体pPICZαA-GDH的构建 将本实验室保藏的重组质粒pMD-GDH和pPICZαA分别进行EcoRⅠ和NotⅠ双酶切,胶回收目的片段,酶切产物利用T4 DNA连接酶进行连接,转化到E.coliTop10。酶切鉴定阳性克隆,并送至中科希林生物科技有限公司进行测序。
1.2.2 PCR点突变 由于FAD-GDH基因序列有BglⅡ酶切位点,不利于后期同尾酶法构建基因多拷贝菌株,所以需要设计引物将1 404位点由A突变为G。引物序列为A-G:F(ATCAGAAGGTCTTTCGAGTCTTACCCTTTG);A-G:R(GAAA-GACCTTCTGATGTACTTAGCAACAGCA)。用Q5连接酶进行PCR扩增,程序:98 ℃,5 min;98 ℃,30 s,退火温度分别设为58、60、62 ℃,60 s,72 ℃,延伸10 min,20个循环;72 ℃,延伸20 min;16 ℃保存。模板用之前构建的pPICZαA-GDH质粒。PCR结果用DpnⅠ酶消解,热激转化Top10感受态,分别涂布于LB+Zeocin平板上,37 ℃过夜培养。分别挑取单菌落于LB液体培养基中培养,提取质粒,进行BglⅡ和BamHⅠ双酶切验证,酶切正确的送通用生物(安徽)股份有限公司测序,进一步验证确认。
1.2.3 FAD依赖的葡萄糖脱氢酶基因的多拷贝表达载体的构建 多表达盒载体的构建如图1所示。具体如下:质粒pPICZαA-GDH经BglⅡ、BamHⅠ双酶切,回收表达框,与经BamHⅠ单酶切、rSAP去磷酸化回收质粒酶连,将连接产物转化至大肠埃希菌感受态Top10中,筛选阳性克隆子后进行试剂盒提质粒,即得到FAD依赖的葡萄糖脱氢酶两拷贝重组表达载体pPICZαA-2GDH。将两拷贝重组表达载体pPICZαA-2GDH分别进行BglⅡ、BamHⅠ双酶切和BamHⅠ单酶切、rSAP去磷酸化,然后分别将酶切产物切胶回收,将表达框片段和线性化的重组表达载体片段用 T4 DNA 连接酶连接,将连接产物转化至大肠埃希菌感受态中,筛选阳性克隆子,即得到4拷贝的重组表达载体pPICZαA-4GDH。两拷贝载体单切线性化与单拷贝的表达框连接得到3拷贝的重组表达载体pPICZαA-3GDH。

图1 2拷贝载体构建Fig.1 Vector construction of 2-copy
1.2.4 多拷贝酵母表达菌株的构建及验证 将构建成功的不同拷贝数重组载体质粒进行BamHⅠ线性化后分别电转表达宿主PichiapastorisX33感受态,构建重组菌株,电转条件:2 kV,4~5 ms,快速加入1 mL 预冷的YPDS液体培养基,然后置于30 ℃培养箱静置培养2~6 h, 4 000 r/min离心5 min,弃上清,用1 mL生理盐水洗3次,然后将菌液涂布在100 μg/mL Zeocin的YPD平板上,30 ℃培养2~3 d,获得转化子。首先采用平板褪色筛选方法对转化子进行初筛。用无菌牙签将YPD平板上长出来的转化子,复制到具有相同编号的MM和MD平板上,将平板置于28 ℃培养箱培养,24 h后取出,将显色液倾倒于MM平板上,室温10~15 min即可发生褪色反应,阳性菌株在平板上可以将DCIP的蓝色退去,出现褪色圈,据此确定阳性率,根据褪色圈大小初步定性确定酶活大小。根据褪色反应筛选出阳性克隆,每种菌株随机挑选3个转化子,提取基因组DNA,利用实时定量PCR(qPCR)的方法检测各转化子中目的基因的拷贝数[21-22]。qPCR所用引物见表1。选择GAPDH为内参基因。
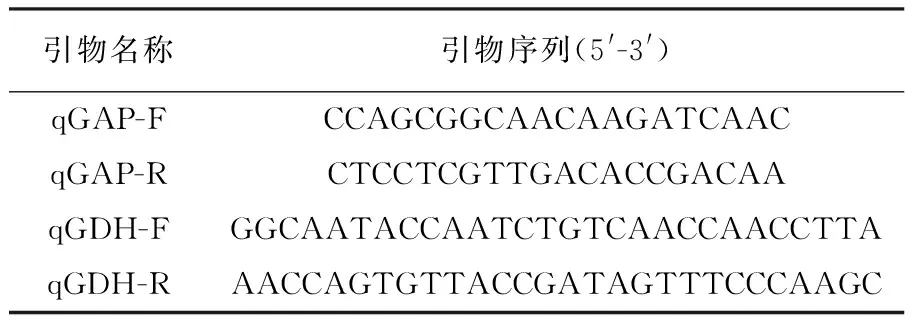
表1 qPCR所用引物 Table 1 The primers used in this study
1.2.5 重组蛋白的诱导表达及相关分析 随后对平板初筛得到的转化子进行甲醇诱导表达,测定酶活,具体如下:从MD平板上挑取相应单菌落接种于5 mL 添加100 μg/mL Zeocin的YPCS试管培养基中,14~18 h后加 1%(体积分数)甲醇,之后24 h和48 h后均各加 1%(体积分数)甲醇诱导,72 h收菌,8 000 r/min离心5 min收集上清,测定酶活。至少重复3次。FAD依赖葡萄糖脱氢酶的酶活测定,使用DCIP 作为电子受体,30 ℃反应180 s,测定520 nm的吸光值变化[6]。分析比较不同基因拷贝数重组菌株的酶活。
1.2.6 10 L发酵罐放大表达FAD-GDH 选择酶活高且稳定的1拷贝菌株和4拷贝菌株进行10 L罐扩大培养,分别挑单菌落接种于YPD培养基培养10~14 h,按3%(体积分数)接种量转接于100 mL BMGY培养基至OD600=6~8 ,以10%(体积分数)发酵体积接种于已灭菌的4.5 L BSM培养基中开始发酵培养。甲醇诱导期间每隔24 h加VC水溶液(终浓度0.45 μmol/L),30 ℃培养,每12 h 取样测定发酵液酶活;甲醇诱导72 h后外源添加VB2(终浓度0.5 mmol/L),测定上清液酶活,比较FAD-GDH蛋白质的表达。发酵具体方法参照文献[23-24]。
2 结果与分析
2.1 单拷贝重组表达载体的构建与鉴定
重组质粒pMD-GDH和购买的载体pPICZαA经过EcoRⅠ 和NotⅠ 双酶切结果如图2所示,酶切完全,大小正确。
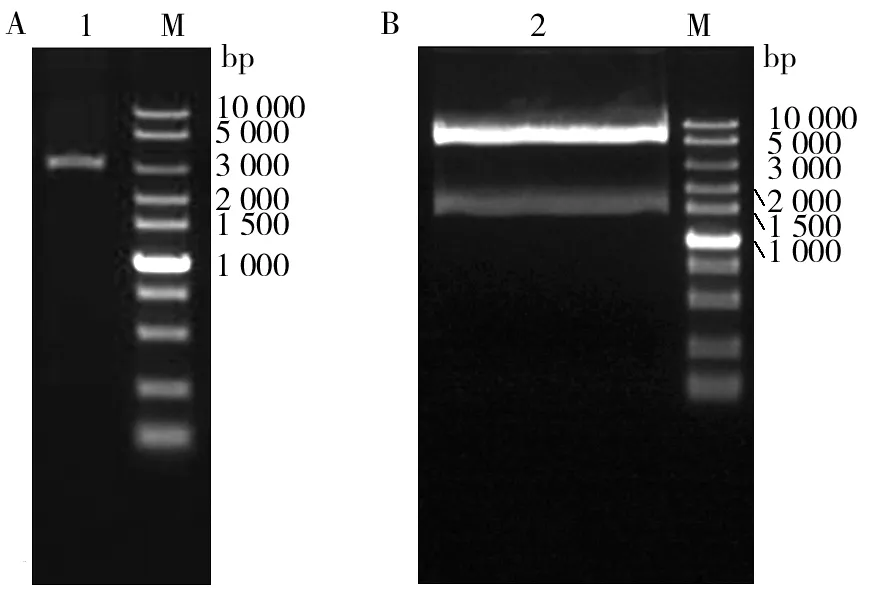
图2 pPICZαA(A)和pMD-GDH(B)双酶切结果Fig.2 Double digestion results of pPICZαA(A) and pMD-GDH(B) M:DNA分子量标准;1:经EcoRⅠ 和NotⅠ 双酶切后的pPICZαA;2:经EcoRⅠ 和NotⅠ 双酶切后的pMD-GDHM:DNA ladder;1: pPICZαA digested with EcoRⅠ and Not I; 2:pMD-GDH digested with EcoRⅠ and NotⅠ
连接产物转化到E.coliTop10,并在含Zeocin的低盐LB平板上筛选阳性菌落。提取阳性菌落质粒,经EcoRⅠ和NotⅠ双酶切验证,得到1.7 kb和3.6 kb两个片段,亦与预期大小相符(图3),说明构建成功。以验证正确的质粒为模板进行点突变,经测序和BglⅡ和BamHⅠ 双酶切验证(图3),获得突变正确的pPICZαA-GDH表达载体,用于后期多拷贝载体构建。
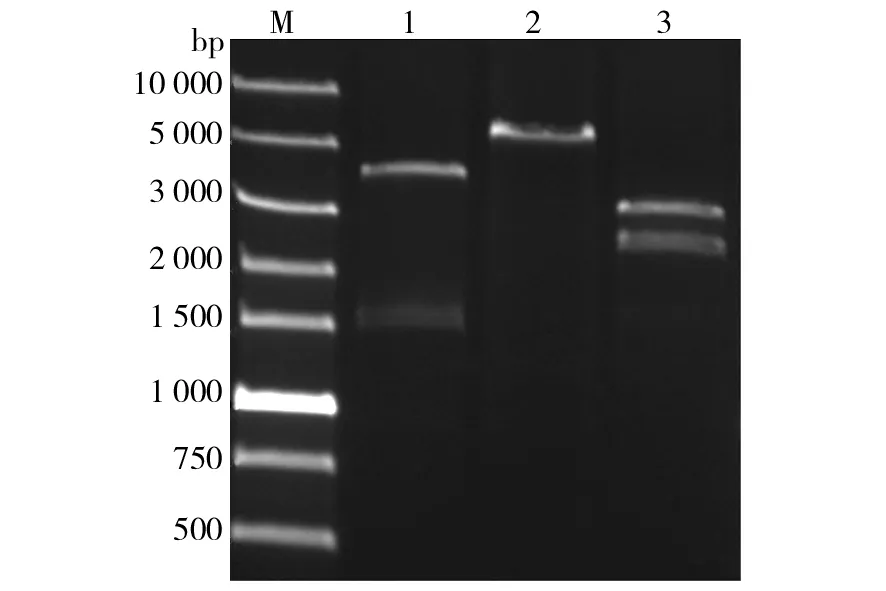
图3 重组质粒pPICZαA-GDH酶切验证结果Fig.3 Enzyme cleavage results of pPICZαA-GDHM:DNA分子量标准;1:经EcoRⅠ 和NotⅠ 双酶切后的pPICZαA-GDH;2:经BamHⅠ单酶切后的pPICZαA-GDH;3:经BglⅡ 和 BamHⅠ双酶切后的pPICZαA-GDHM:DNA ladder; 1:pPICZαA-GDH digested with EcoR I and NotⅠ; 2:pPICZαA-GDH digested with BamHI; 3:pPICZαA-GDH digested with BamH Ⅰ and BglⅡ
2.2 多拷贝重组表达载体的构建与鉴定
首先,将构建的重组表达质粒 pPICZαA-GDH分别用BamHⅠ单酶切、rSAP去磷酸化,BglⅡ和BamHⅠ双酶切得到单拷贝的表达盒GDH,通过胶回收获取两段目的片段,进行T4连接酶连接,然后将连接产物转化至E.coliTop10感受态细胞中,对获得的转化子使用BglⅡ和BamHⅠ做酶切验证,通过筛选获得2拷贝阳性克隆子pPICZαA-2GDH。再将2拷贝表达载体用BglⅡ和BamHⅠ双酶切,回收2拷贝表达框,与2拷贝表达载体单酶切骨架酶连,得到4拷贝表达载体pPICZαA-4GDH。再将1拷贝表达框与2拷贝表达载体单酶切骨架酶连,得到3拷贝表达载体pPICZαA-3GDH。不同拷贝数载体BglⅡ和BamHⅠ双酶切结果如图4所示,1、2、3、4拷贝质粒分别产生3.4、6.7、10、10.3 kb的片段,表明多拷贝表达盒表达载体构建成功。

图4 多拷贝表达载体酶切电泳分析图Fig.4 Analysis of constructed plasmids with restriction endonucleases, digested with BglⅡ and BamHⅠM:DNA分子量标准;1~4:经BglⅡ 和 BamHⅠ双酶切后的pPICZαA-GDHn(n=1,2,3或4)M:DNA ladder;1-4: pPICZαA-GDHn(n=1,2,3 or 4) digested with BamHⅠ and BglⅡ
2.3 多拷贝酵母表达菌株的构建及验证
将验证正确的不同基因拷贝数表达载体pPICZαA-GDH、pPICZαA-2GDH、pPICZαA-3GDH和pPICZαA-4GDH用BamH Ⅰ线性化,电击转化表达宿主PichiapastorisX33,然后再涂布于抗性平板上得到酵母转化子。
通过显色法快速初步筛选阳性转化子,加入显色液5、10、15 min的显色结果如图5所示,刚加入显色液时平板呈现蓝色,菌株周围开始有褪色圈,但不明显;10 min后平板呈现紫红色,阳性菌株周围的褪色圈变大,15 min后褪色更明显。其中非阳性转化子没有褪色反应(图5中箭头所指菌株)。

图5 显色法筛选结果Fig.5 Chromogenic screening results
然后挑取阳性转化子进行试管诱导发酵,用1%甲醇诱导72 h,测定上清液FAD依赖葡萄糖脱氢酶活,并以此来筛选高酶活菌株。通过qRT-PCR检测了阳性转化子中的基因拷贝数,结果表明重组质粒都成功整合到毕赤酵母的基因组上,目的基因拷贝数符合预期(表2)。

表2 酵母转化子的拷贝数分析 Table 2 The copy number analysis of the yeast transformants
从所有保存菌株中挑选1、2、3和4拷贝各3株,重复诱导3次,并对1、2、3和4拷贝的重组酵母菌株进行表达水平的比较。结果表明,在试管水平,单拷贝平均酶活11.953 U/mL,2拷贝平均酶活27.99 U/mL,3拷贝平均酶活44.792 U/mL,4拷贝平均酶活57.027 U/mL;分别比单拷贝提高2.34、3.75、4.78倍(图6)。由此可见,多拷贝表达载体的构建可以提高FAD依赖的葡萄糖脱氢酶的表达水平。

图6 试管水平不同基因拷贝数重组菌株酶活比较Fig.6 Comparison of enzyme activities of recombinant strains with different gene copy numbers at test tube level
2.4 10 L发酵罐扩大培养
选择酶活高且稳定的1拷贝菌株和4拷贝菌株进行10 L罐扩大培养,结果如图7所示。1拷贝菌株诱导108 h酶活达到最高697.125 U/mL(图7A),4拷贝菌株诱导132 h酶活达到最高1 063.279 U/mL(图7B);OD600和湿重均随着诱导时间逐步增加。
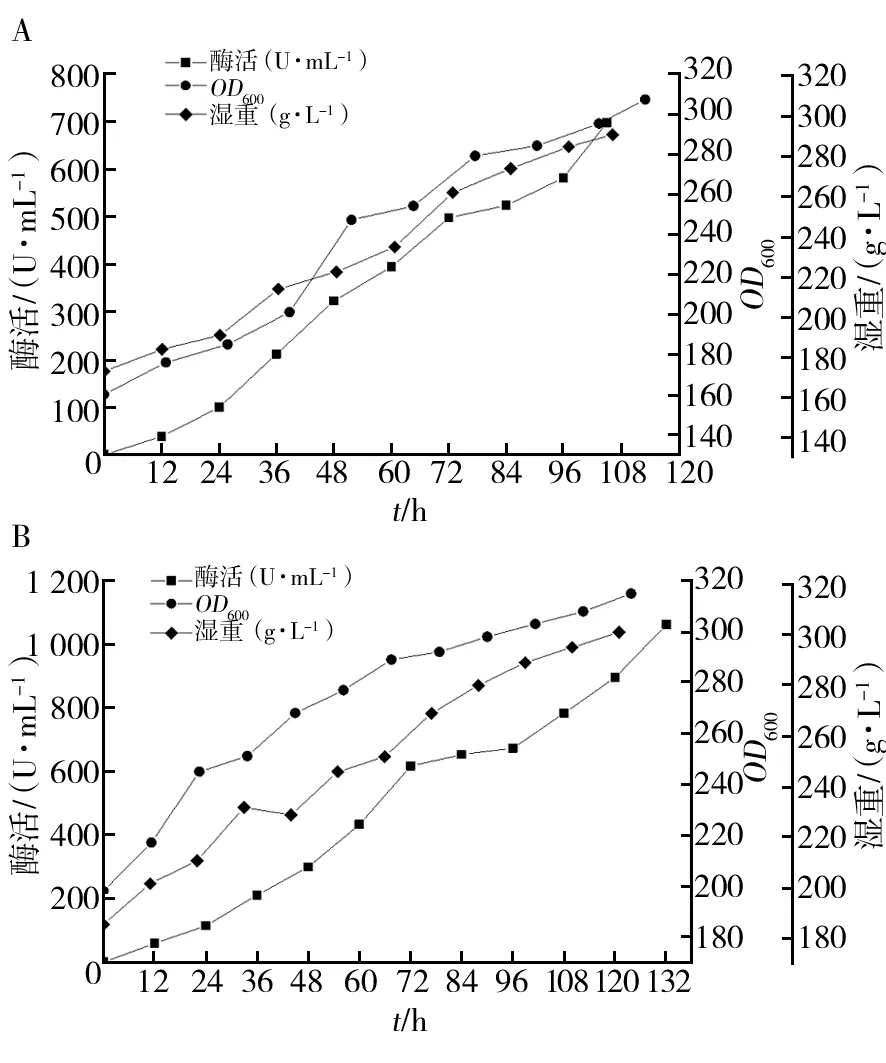
图7 10 L发酵罐扩大培养结果Fig.7 Culture results of 10 L Fermentation A:X33/pPICZαA-GDH菌株酶活生长曲线;B:X33/pPICZαA-4GDH菌株酶活生长曲线A:Growth curve of X33/pPICZαA-GDH; B:Growth curve of X33/pPICZαA-4GDH
从图8可以看出,甲醇诱导前60 h二者酶活差异不大,60 h以后4拷贝菌株酶活一直比1拷贝菌株酶活高。4拷贝菌株在诱导72 h后酶活617.3 U/mL,是原试管水平发酵72 h(57.027 U/mL)时的10.82倍。
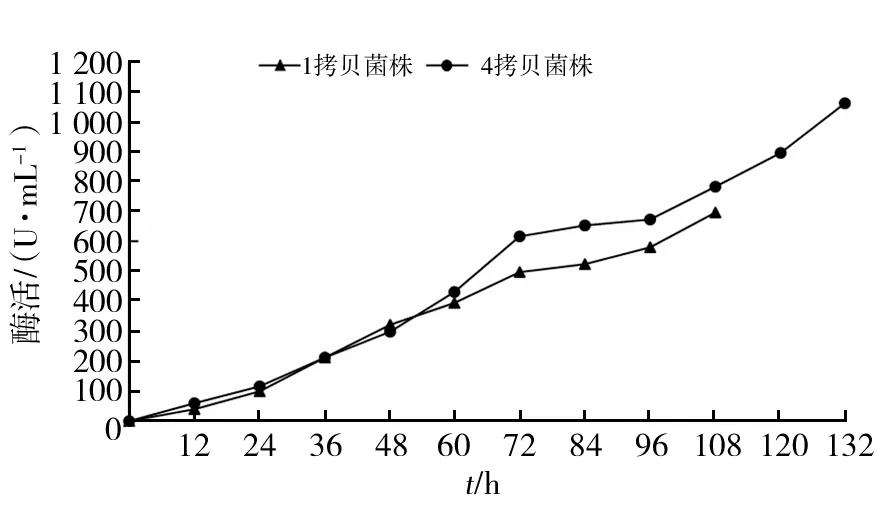
图8 10 L发酵罐上清液酶活比较Fig.8 Comparison of enzyme activity of 10 L Fermentation
3 讨 论
FAD依赖的葡萄糖脱氢酶作为血糖检测用酶有广阔的应用前景,但其在天然宿主菌中表达量很低,在大肠埃希菌进行重组表达时易形成包涵体,不易分离[25]。目前主要依赖进口,还没有实现国产化。前期研究构建的含单拷贝表达盒载体的重组毕赤酵母菌株X33/pMD-GDH产量还不能满足生产需求,亟需利用基因工程技术实现高效表达。近年来,有很多通过同尾酶法构建基因多拷贝表达盒载体提高外源基因在毕赤酵母中表达量的报道。表达盒是独立的顺式作用元件,多个表达盒可以同向连接在同一载体中,转化宿主后实现多个基因的共表达[26]。所以,本研究拟通过同尾酶法构建含不同拷贝表达盒载体pPICZαA-nGDH的重组菌株,使每一个表达盒都可以独立表达目的蛋白,以提高GDH的表达量。通过qRT-PCR证明,重组菌株所含载体的表达盒数与GDH基因的拷贝数之间存在正相关关系。
近年来有研究发现,基因拷贝数的增加与外源重组蛋白的表达量并不总是正相关关系,有时候基因拷贝数对外源蛋白的表达会起负效应。这与外源蛋白表达受很多因素影响有关,例如不同启动子、毕赤酵母的不同表型、不同分泌信号和表达机制等[27]。Cos等[28]为提高根霉脂肪酶表达量构建了Muts和Mut+型重组表达毕赤酵母多拷贝菌株,发现二者产量相对于单拷贝的产量都有所下降,分别降低了5%和30%。Song等[29]构建了不同拷贝谷氨酰转胺酶基因mtg的表达菌株,发现基因拷贝数从1增加到3时,表达量增加,但是拷贝数再增加后菌株的酶活明显下降。Hohenblum等[30]在pGAP启动子调控下将胰岛素酶基因拷贝数从1增加到3,但是其表达量没有变化。
同时,通过同尾酶法增加拷贝数也受到其他因素的限制。比如,表达盒插入方向随机、载体自连、载体长度逐渐增加等因素均会降低构建多拷贝表达载体的效率。综上,本研究只构建了1~4拷贝表达盒载体。焦梁成[31]用同尾酶法最高成功构建4拷贝米根脂肪酶ROL重组载体,而用改进的生物砖法较容易构建获得含8拷贝表达盒重组载体。因此,进一步研究将探索更多的方法提高表达量。
本研究用按照毕赤酵母偏好性进行优化的FAD依赖的葡萄糖脱氢酶基因序列与pPICZαA质粒通过同尾酶法构建了含1~4拷贝表达盒表达载体,将其电击转入PichiapastorisX33,获得FAD依赖的葡萄糖脱氢酶高效分泌表达重组菌株。qRT-PCR测定结果表明重组菌株所含载体的表达盒数与FAD-GDH基因的拷贝数之间存在正相关关系。试管水平,重组菌诱导发酵72 h,酶活达到最高,其中4拷贝的转化子表达水平最高;选择1拷贝和4拷贝转化子进行10 L发酵罐扩大培养,1拷贝菌株诱导108 h,酶活达到最高697.125 U/mL,4拷贝菌株诱导132 h,酶活达到最高1 063.279 U/mL,比1拷贝酶活提高52.52%。结果表明通过增加目的基因拷贝数策略有助于提高FAD-GDH的表达量,为其进一步扩大生产奠定理论和技术基础,使其能够更好地应用于血糖检测等方面。
猜你喜欢
河北医学(2021年10期)2021-10-27
中国临床医学影像杂志(2019年6期)2019-08-27
中国生殖健康(2018年1期)2018-11-06
中国调味品(2017年2期)2017-03-20
创新作文(小学版)(2016年16期)2016-11-11
现代检验医学杂志(2016年5期)2016-08-20
中国科技信息(2015年2期)2015-11-16
计算机与网络(2015年12期)2015-06-21
发明与创新(2015年25期)2015-02-27
医学综述(2014年24期)2014-03-08
