
异尾次目寄居蟹总科线粒体基因组比较分析及系统发育研究
2024-02-07 07:29江婷琪胡璧麟张楠楠汪凯欣吕振明
水生生物学报 2024年2期
江婷琪 胡璧麟 张楠楠 汪凯欣 吕振明 龚 理
(浙江海洋大学海洋科学与技术学院, 海洋生物种质发掘与利用国家地方联合工程研究中心, 舟山 316022)
异尾次目(Anomura)隶属于节肢动物门(Arthropoda)软甲纲(Malacostraca)十足目(Decapoda), 是一类体型介于虾类和蟹类之间高度特化的甲壳动物。根据形态特征, 异尾次目可分为铠甲虾型(Squat lobster form)、蟹型(Crab-like form)和寄居蟹型(Hermit form)三个主要类群; 其中, 寄居蟹型又可以分为对称寄居蟹型(Symmetrical hermit form)和不对称寄居蟹型(Asymmetrical hermit form)。对称寄居蟹型常见于门螯寄居蟹科(Pylochelidae)种类, 不对称寄居蟹型多见于陆寄居蟹科(Coenobitidae)、拟寄居蟹科(Parapaguridae)和寄居蟹科(Paguridae)绝大多数种类。寄居蟹独特的形态特征代表了甲壳动物从长尾类(Macrura)到短尾类(Brachyura)的过渡状态, 因而在甲壳动物进化过程中占据十分重要的地位[1]。
虽然1802年Latreille就建立了寄居蟹总科(Paguridea), 但是该总科分类系统长期以来未有定论[2,3]。最初的研究把所有寄居蟹都归于寄居蟹总科, 直至1957年, MacDonald等[4]提出将寄居蟹分为寄居蟹总科和陆寄居蟹总科, 其中寄居蟹总科包括石蟹科(Lithodidae)、拟寄居蟹科和寄居蟹科, 陆寄居蟹总科包括陆寄居蟹科和活额寄居蟹科(Diogenidae),该分类体系并未考虑澳洲寄居蟹科(Lomisidae) 的归属。McLaughlin[5]则认为澳洲寄居蟹科应该归属独立总科, 同时提议废除陆寄居蟹总科, 仅保留寄居蟹总科。Martin和Davis[6]支持仅设寄居蟹总科的观点, 并新增门螯寄居蟹科。McLaughlin等[7]基于形态特征重建了寄居蟹总科系统发育树, 提议将石蟹科独立出来, 并将其归于石蟹总科(Lithodoidea)。当前, 石蟹科到底归于寄居蟹总科还是石蟹总科仍有争议。由于螯盖寄居蟹科(Pylojacquesidae)物种稀少(目前仅发现2属2种), 直至2001年该科才被建立。至此, 寄居蟹总科包括拟寄居蟹科、寄居蟹科、陆寄居蟹科、活额寄居蟹科、门螯寄居蟹科和螯盖寄居蟹科6科的分类系统正式确立。
虽然, 目前寄居蟹总科的分类系统被绝大多数学者所认可[8—10], 但是该类群内部分类及亲缘关系仍具争议。Richter和Scholz[3]首次基于形态特征分析了寄居蟹类的进化关系, 认为寄居蟹总科中除了最原始的门螯寄居蟹是并系群外, 其余种类均为单系群; 而当前绝大多数分子数据都支持活额寄居蟹科为并系群[11,12]。随着寄居蟹科分子数据不断增加, 越来越多的研究发现该科也为非单系群, 如寄居蟹科中的Pagurus longicarpus不与同属物种聚为一支, 而是与寄居蟹科和石蟹科组成的姐妹支聚支[10,13],支持寄居蟹科的非单系性。此外, 寄居蟹和石蟹之间的进化关系也是甲壳动物系统进化学家关注的焦点[8,14,15]。因此, 开展寄居蟹总科系统分类学研究对异尾次目乃至整个甲壳类分类及系统演化具有重要意义。
线粒体基因组被广泛应用于分子系统学研究,解决了很多长期以来备受争议的分类鉴定及系统进化等问题。如传统形态学认为沙蟹总科和方蟹总科是单系群, 但是越来越多的分子结果(包括线粒体基因组数据)显示这两个总科为并系群[16—19]。除了线粒体基因组序列本身外, 也有研究发现线粒体基因排序也可以为分子系统学提供有用信息。如Tan等[20]比较了已有异尾次目和短尾次目线粒体全序列后提出基因重排可以用于异尾次目系统发育研究, 如前期基于线粒体基因组序列构建的系统发育树无法确定辉虾属(Aegla)(辉虾总科)和澳洲寄居蟹属(Lomis)(澳洲寄居蟹总科)之间的亲缘关系,但是基因排序信息则显示澳洲寄居蟹总科和柱螯虾总科有着最近的亲缘关系, 两者再与辉虾总科形成姐妹群关系, 随后不断有研究证实了这一观点[12,13]。关于线粒体基因重排现象, 目前主要用串联复制-随机丢失(Tandem duplication and random loss)[21]、串联复制-非随机丢失(Tandem duplication and nonrandom loss)[22]和线粒体内重组(Intramitochondrial recombination)[23]等模型假说来解释。
然而, 相比甲壳类其他类群, 研究人员对异尾次目线粒体基因组关注度明显不足; 尤其是寄居蟹总科, 截2023年4月, GenBank数据库中仅有16个物种的线粒体基因组全序列, 这极大阻碍了寄居蟹分子系统学研究发展。因此, 本研究以我国海域分布最广泛的活额寄居蟹科物种作为研究对象, 以刺足真寄居蟹(Dardanus hessii)为代表种, 首次测定其线粒体基因组全序列, 并结合已公布的16个寄居蟹物种的基因组全序列, 对寄居蟹总科线粒体基因组序列特征及基因排序进行了比较分析, 同时构建了寄居蟹总科系统发育树, 探究线粒体基因重排在系统进化研究中的适用性。本研究结果不仅丰富了寄居蟹总科的线粒体基因组数据, 同时为异尾次目分类及系统进化提供新的观点和思路。
1 材料与方法
1.1 样品采集及测序
刺足真寄居蟹样品采自浙江省舟山市桃花岛,参考《中国海活额寄居蟹科分类学研究》[24]进行形态学鉴定。取约100 g肌肉组织利用二代测序技术进行线粒体基因组全序列测定(上海元莘生物医药科技有限公司)。测序前先使用插入片段大小为400 bp的VAHTS Universal Plus DNA文库制备试剂盒构建基因文库, 在Illumina NovaSeq 6000高通量测序平台进行PE150测序; 使用NOVO Plasty[25]软件对去除接头的有效数据(Clean data)进行从头组装; 将鳞纹真寄居蟹线粒体基因组(GenBank登录号: MW147148)作为种子序列进行序列延伸。为了评估二代测序的准确性, 研究还利用一代测序技术(Sanger测序)测定了线粒体COⅠ基因序列, 比较两种测序方法得到的序列相似性。
1.2 线粒体基因组注释和序列分析
基于MITOS Web Server[26]及tRNAscan-SE 1.21[27]预测结果, 利用Sequin软件(version 15.10, http://www.ncbi.nlm.nih.gov/Sequin/)对新组装的刺足真寄居蟹线粒体基因组进行注释; 使用NCBI-BLAST对蛋白质编码基因和核糖体RNA基因进行边界分析; 控制区由于序列变异较大, 根据近缘种序列及其相邻基因位置判断。利用以下公式计算碱基的链不对称性: GC-skew=(G-C)/(G+C); AT-skew=(A-T)/(A+T)[28]。使用 MEGA X软件[29]计算核苷酸含量。从GenBank 数据库中下载另外16种寄居蟹总科物种的线粒体COⅠ基因序列, 结合本研究测定的刺足真寄居蟹物种COⅠ序列, 利用软件Clustal X 2.0[30]对这些序列进行比对, 并基于Kimura 2-parameter (K2P)参数替代模型计算17个物种COⅠ的遗传距离, 利用软件MatGAT 2.02[31]进行多序列相似性比较分析。
1.3 线粒体基因组比较分析
以刺足真寄居蟹线粒体基因组作为参考序列,用CGView Comparison Tool (CCT)[32]软件对所有17种寄居蟹总科物种线粒体基因组序列进行比较,同时用Mauve v2.4.0[33]软件对其进行共线性分析。采用CREx[34]软件预测基因重排事件, 基因重排包括倒置(R)、移位(T)和倒置移位(RT)三种类型。
1.4 系统发育分析
从GenBank数据库下载目前已有16种寄居蟹总科物种线粒体基因组全序列, 加上本研究新测定的刺足真寄居蟹基因组, 以方蟹总科的侧足厚蟹(Helice latimera)和伍氏拟厚蟹(Helicana wuana)为外类群, 基于13个蛋白质编码基因, 使用PhyloSuite[35]同时构建寄居蟹总科最大似然树(ML)和贝叶斯树(BI)。最大似然树使用IQ-TREE构建, 其中bootstrap设置为Ultrafast, Num of bootstrap设置为100000,抽样次数为1000。贝叶斯树使用MrBayes构建, 使用4条蒙特卡罗马尔可夫链(Markov Chain Monte Carlo, MCMC)同时运行2000000代, 抽样频率为1000, 摒弃25%的老化样本。
2 结果
2.1 刺足真寄居蟹线粒体基因组序列特征
刺足真寄居蟹线粒体基因组全序列长度为17821 bp(GenBank登录号OP930882), 包含37个基因(13个蛋白质编码基因, 22个tRNA和2个rRNA基因)和1个控制区。13个蛋白质编码基因总长为11183 bp, 共编码3716个氨基酸。所有蛋白质编码基因均以典型的ATN作为起始密码子; 除ND5和ND6基因以T作为终止密码子外, 其余蛋白质编码基因以TAA或TAG作为终止密码子(表1)。tRNA基因分散在整个线粒体基因组中, 22个tRNA的总长度为1471 bp。16S rRNA位于ND1和tRNA-Val之间, 长度为1393 bp;12S rRNA位于tRNA-Val和CR之间, 长度为798 bp。虽然线粒体基因组各基因排列非常紧凑, 但是在刺足真寄居蟹基因组中23处共发现270 bp的基因间隔区, 其中最长的为75 bp, 介于tRNA-His和ND4之间。当然, 同时也在5处发现了累计15 bp的重叠区域, 最长的重叠区为7 bp, 位于ND4和ND4L之间(表1)。
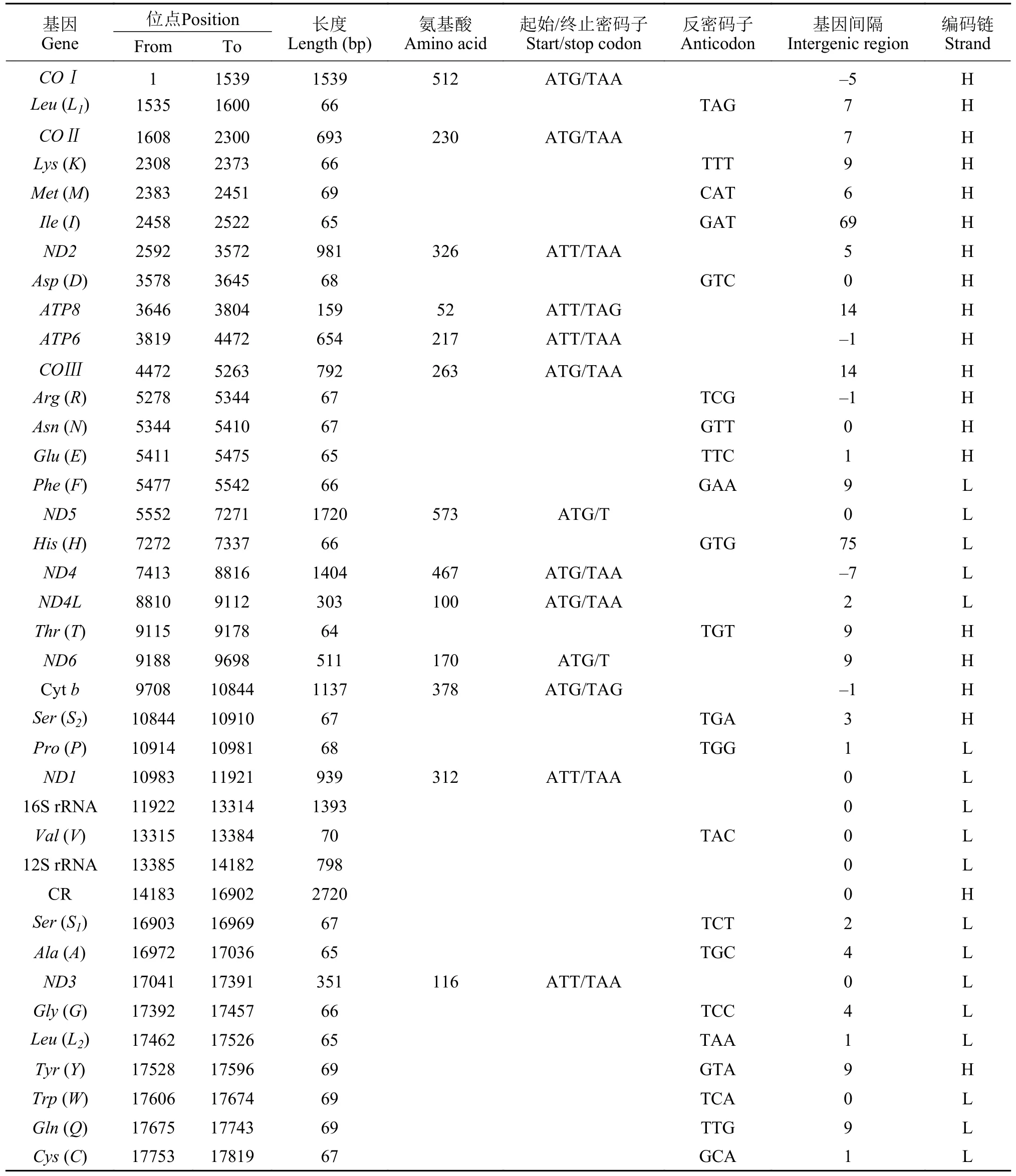
表1 刺足真寄居蟹线粒体基因组特征Tab.1 Features of the mitochondrial genome of D.hessii
2.2 寄居蟹总科物种线粒体基因组比较分析
用CCT软件对17种寄居蟹总科的线粒体基因组进行了比较分析, 结果显示, 17个物种的线粒体基因组都发生了基因重排, 且基因排序和序列相似性都相对保守。除控制区和16S rRNA部分序列有较大变异外, 其余基因相似性普遍高于82%, 尤其是COⅠ和COⅢ基因在所有寄居蟹物种线粒体基因组中十分保守(图1)。
COⅠ基因作为最常用的DNA条形码, 在物种分类鉴定有着广泛的应用。因此本研究计算了17种寄居蟹总科物种的COⅠ基因遗传距离及其序列相似性。结果显示, 日本寄居蟹(Pagurus japonicus)和长腕寄居蟹(Pagurus filholi)间的遗传距离最小(0.005), 二者对应的序列相似性也最高(99.5%), 表明两者很有可能为同一物种; 最大的遗传距离出现在Pagurus longicarpus和灰白陆寄居蟹(Coenobita rugosus)之间(0.292), 对应的序列相似性为75.7%。本研究新测定的刺足真寄居蟹和同属的红星真寄居蟹(Dardanus asperses)及鳞纹真寄居蟹(Dardanus arrosor)的遗传距离最小, 分别为0.203和0.204, 序列相似性分别为82.1%和82.0%。
研究还对17种寄居蟹总科物种的线粒体基因组共线性进行了比较分析, 软件分析显示, 所有寄居蟹物种线粒体基因组均可分为5个大的保守区域(图2, A—E), 其中区域A(P-ND1-16S rRNA) 的位置和长度在所有寄居蟹物种线粒体基因组中都是最保守的, 区域B的位置和长度变异最大, 其余3个区域则相对保守。值得注意的是毛氏门螯寄居蟹(Pylocheles mortensenii) 基因组中的区域B(12S rRNA)发生了倒置, 由轻链编码变成了重链编码, 而在其余16种寄居蟹总科物种线粒体基因组均仍由轻链编码; 此外, 区域E (T-ND6-Cytb-S2) 在红星真寄居蟹基因组中发生了较明显的移位, 而其在其余16种寄居蟹总科物种线粒体基因组中位置十分保守。
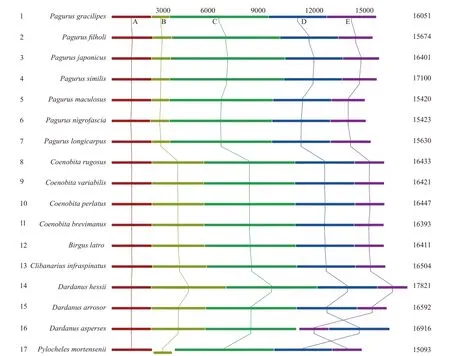
图2 17种寄居蟹总科物种线粒体基因组共线性分析Fig.2 Collinearity analysis of 17 Paguroidea mitogenomes
进一步分析发现17种寄居蟹总科物种的线粒体基因组共分为4种共线性类型, 其中7种寄居蟹科物种共享一种共线类型(1—7); 5种陆寄居蟹科和3种活额寄居蟹科物种共享一种共线类型(8—15);活额寄居蟹科的红星真寄居蟹独享一种共线类型(16), 与该科其他3个物种线粒体基因排序明显不同, 表现在红星真寄居蟹基因组中T-ND6-Cytb-S2移位至R-N-E基因簇下游, 而在5种陆寄居蟹和3种活额寄居蟹基因组中, 该区域移位至ND4L下游;门螯寄居蟹科的毛氏门螯寄居蟹独享一种共线类型(17), 该类型与其他3种类型最大的区别是12S rRNA发生了倒置。
2.3 线粒体基因重排分析
与泛甲壳动物(Pancrustacea) 线粒体基因组原始基因排序(Type 0)相比, 刺足真寄居蟹基因顺序经历了大规模的重排, 共有6处基因或基因簇位置发生了显著变化, 涉及到12个tRNA基因(L2、G、A、S1、P、L1、I、Q、M、W、C和Y)和2个蛋白质编码基因(ND3和ND2)。在这6处重排中, 包含了倒置移位(RT)和移位(T)两种重排类型。单个的L2和L1基因、G-ND3-A-S1和W-C-Y基因簇发生了倒置移位, 单个的P基因和I-Q-M-ND2基因簇仅发生移位,未发生倒置; 其中L2基因由重链编码倒转至轻链,同时从COⅠ基因下游移至G基因的下游(图3 ①);G-ND3-A-S1基因簇也是由重链编码倒转至轻链, 变成了S1-A-G-ND3排序, 位置从COⅢ基因的下游移至了控制区(CR)的下游(图3 ②);P基因未发生倒置, 仅发生位置上的变化, 从T基因的下游移至S2基因的下游(图3 ③);L1基因则由轻链编码倒转至重链, 位置从ND1基因的下游移位至COⅠ基因的下游(图3 ④);I-Q-M-ND2基因簇仅位置发生变化, 其被分为两部分, 其中I-M-ND2整体移至K基因的下游, 且I和M的前后位置发生了互换, 形成了M-IND2排序,Q基因则移至线粒体基因组的线性末端位置(图3 ⑤); 最后一处重排发生在W-C-Y基因簇,其中W和Y基因发生了倒置移位, 形成了新的Y-WC排序(图3 ⑥)。
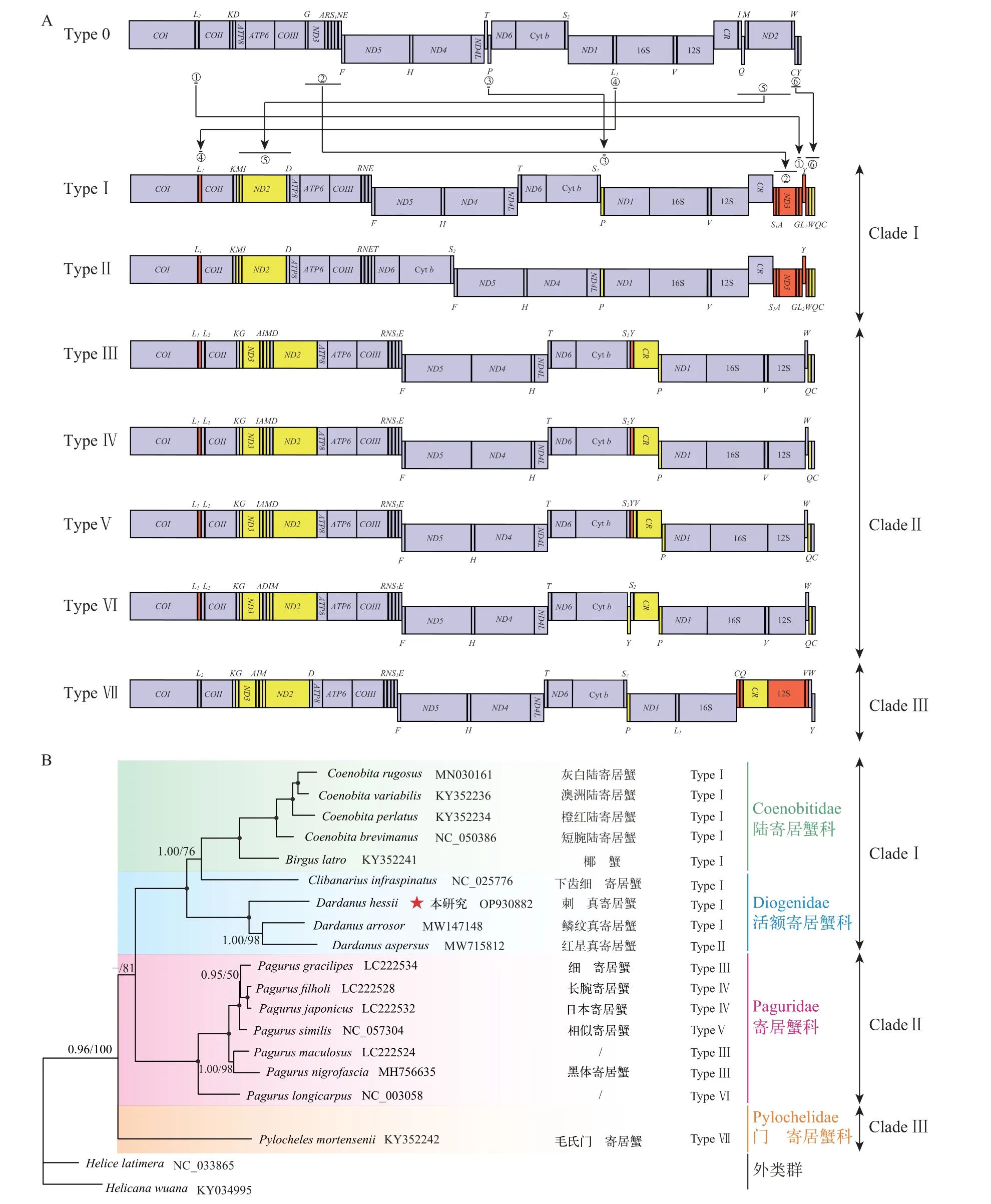
图3 寄居蟹总科物种线粒体重排类型(A) 及寄居蟹总科系统发育关系(B)Fig.3 Mitochondrial rearrangement types of species of Paguridea (A) and phylogenetic relationship of Paguridea (B)
研究进一步比较了所有寄居蟹总科物种线粒体基因排序, 结果显示17种寄居蟹总科物种线粒体基因组表现出7种不同类型的基因重排, 其中寄居蟹科物种线粒体基因组贡献了4种基因重排类型(图3Ⅰ—Ⅳ); 所有陆寄居蟹科物种和3种活额寄居蟹科共享一种重排类型(图3Ⅴ); 活额寄居蟹科的红星真寄居蟹和门螯寄居蟹科的毛氏门螯寄居蟹各自独享一种重排类型(图3Ⅵ和Ⅶ)。相比其余3种基因排序(图3Ⅴ—Ⅶ) 之间的差异, 寄居蟹科中4种基因重排类型(图3Ⅰ—Ⅳ) 内部差异显得很小,仅在AIMD基因簇排序、H及V的位置上有差异。活额寄居蟹科具有两种不同的基因重排类型(图3Ⅴ和Ⅵ), 两者之间差异仅体现在T-ND6-Cytb-S2基因簇的位置, 包括大部分活额寄居蟹科在内的寄居蟹总科物种T-ND6-Cytb-S2基因簇位于ND4L基因下游, 仅在活额寄居蟹科的红星真寄居蟹基因组中位于R-N-E基因簇下游。门螯寄居蟹科基因排序(图3Ⅶ)最大的不同在于12S rRNA的编码链, 在其余6种基因重排类型中, 12S rRNA均由轻链编码, 而该基因在门螯寄居蟹科线粒体基因组中发生了倒置, 由轻链编码转为重链编码, 与线粒体基因组共线性分析结果一致。
2.4 系统发育分析
本研究基于13个蛋白质编码基因的核苷酸序列同时构建了寄居蟹总科最大似然树(ML tree)和贝叶斯树(BI tree), 结果显示两种方法构建的系统发育树具有相同的拓扑结构; 因此, 本研究只显示其中一种树, 节点处同时标记出贝叶斯后验概率和最大似然树自展值。系统树显示本研究新测定的刺足真寄居蟹跟鳞纹真寄居蟹及红星真寄居蟹聚为一支, 表明这三者亲缘关系很近, 组成了活额寄居蟹科的一部分; 然而, 活额寄居蟹科的另一个物种, 下齿细螯寄居蟹并没有与同科的3个物种聚在一起, 而是先与陆寄居蟹科聚为一支, 再与3种活额寄居蟹科物种形成姐妹群, 表明活额寄居蟹科为非单系群。寄居蟹总科内部亲缘关系为活额寄居蟹科与陆寄居蟹科形成姐妹群后再与寄居蟹科聚在一起, 最后与门螯寄居蟹科聚支。需要提醒的是,NCBI数据库中长腕寄居蟹(LC222528)与日本寄居蟹(LC222532)的线粒体基因组序列可能来自同一物种, 与COⅠ遗传距离和序列相似性结果一致。两者形态上有较大差异(如楯部比例、螯上小刺或短刚毛等), 暗示其中至少有一个物种鉴定有误。
3 讨论
异尾次目分类及演化是进化系统学备受关注的科学问题。然而, 该类群分类及内部亲缘关系长期以来一直缺乏定论[8,15,36,37]。随着测序技术的飞速发展及测序成本的不断降低, 线粒体基因组全序列在分类鉴定、系统进化和适应性进化等领域解决了很多备受争议的难题[16,20,38]。本研究基于17种寄居蟹总科物种13个线粒体蛋白质编码基因序列重建了寄居蟹总科系统发育关系, 结果显示活额寄居蟹科与陆寄居蟹科亲缘关系最近, 两者形成姐妹群后再与寄居蟹科聚为一支, 最后与门螯寄居蟹科聚在一起; 该聚类关系与绝大多数分子系统树结果一致[12,20,39,40]。虽然, 本研究也支持了活额寄居蟹科的非单系性, 但是值得注意的是造成该科非单系的关键物种, 下齿细螯寄居蟹与陆寄居蟹科的关系与前人研究结果略有区别。在近期的异尾次目系统发育研究中, 陆寄居蟹科先与大多数活额寄居蟹科聚支后再与下齿细螯寄居蟹聚在一起[11], 而本研究并入刺足真寄居蟹后则显示陆寄居蟹科先与下齿细螯寄居蟹聚为一支, 再与大多数活额寄居蟹科聚在一起, 该结果与Tsang等[41]基于5个核蛋白编码基因构建的寄居蟹总科系统发育关系一致。不难发现下齿细螯寄居蟹在系统树中的位置并不完全稳定, 今后可增加活额寄居蟹科和寄居蟹科代表种数量进一步厘清这两个科的亲缘关系。
脊椎动物线粒体基因组排序相对稳定, 发生重排的概率较低。而在无脊椎动物线粒体基因组中,线粒体基因重排现象相对较多。以甲壳动物十足目为例, 目前已发现相手蟹科(Sesarmidae)、溪蟹科(Potamonidae)、宝石蟹科(Mithracidae)、寄居蟹科、陆寄居蟹科和活额寄居蟹科等20余科物种线粒体基因组发生显著重排[16,42,43]。用于解释线粒体基因重排的机制主要有串联复制随机丢失模型、串联复制非随机丢失模型和线粒体重组等[23,44]。本研究中寄居蟹总科线粒体基因组呈现7种不同的基因排序, 涉及倒置移位和移位两种重排类型。线粒体重组是最常用于解释倒置现象的模型, 已广泛用于解释包括鱼类[45]、爬行类[46]、昆虫[47]和甲壳类[10]等在内多种生物类群基因倒置现象。如半滑舌鳎(Cynoglossus semilaevis)线粒体基因组中Q基因由轻链编码转为重链编码, 这是鱼类线粒体基因组中首例基因倒置现象, 研究采用线粒体重组模型对Q基因倒置进行了重排推演[40,42]。本研究中的倒置现象同样也可以用重组模型来解释。如在泛甲壳动物线粒体基因组(Type 0)中为V-12S排序, 两者均由轻链编码; 而在毛氏门螯寄居蟹基因组(TypeⅦ)中变为12S-V排序, 且由轻链编码转为重链编码。可能的重排过程为:V的5′端和12S的3′端同时断裂, 形成一个自由的V-12S片段, 该片段在重新连接时发生链倒置, 变成12S-V排序, 且由原来的轻链编码变为重链。线粒体重组模型在其他物种类似的倒置现象中也得到广泛应用[38,39]。线粒体基因移位是十分常见的基因重排现象, 常用串联复制随机丢失模型和串联复制非随机丢失模型来解释。两种模型最大的区别在于复制后的基因被删除的方式, 前者为随机删除, 后者则取决于基因的转录极性和位置, 其删除后的结果是具有相同极性的基因(由同一链编码的基因)聚在一起。本研究中寄居蟹总科重排基因是散状分布在基因组中, 而不是按相同极性聚在一起, 因而综合重排基因序列特征及CREx软件预测结果, 我们认为串联复制随机丢失模型是用来解释寄居蟹总科线粒体基因组产生大规模基因重排现象最合理的假说。
研究还分析了寄居蟹总科线粒体基因排序跟系统进化之间的关系。寄居蟹科虽然拥有4种不同的基因重排类型(Type Ⅰ—Ⅳ), 但是整科物种还是聚为一支, 形成一个单系群(Clade Ⅰ)。陆寄居蟹科和活额寄居蟹科中3个物种共享同一种重排类型(Type Ⅴ), 它们最先聚为一支, 暗示基因排序一定程度上能够反映系统进化信息[40,43]。但是值得注意的是活额寄居蟹科中鳞纹真寄居蟹先与基因排序具有较大区别的红星真寄居蟹聚类, 两者形成姐妹支后再与具有相同基因排序的刺足真寄居蟹聚在一起, 这又与相同基因排序优先聚类的现象相矛盾。造成该“矛盾”现象的原因是红星真寄居蟹基因重排并未对线粒体基因序列产生大的影响, 因而在基于线粒体序列构建系统发育关系时, 物种亲缘关系并未按基因重排类型进行聚类。因此, 基因排序是否适用于系统进化分析应当视具体情况而定;即当基因重排会对序列本身产生较大影响时, 重排信息可能会成为一种理想的分子标记; 反之基因重排可能不适用于系统进化分析。
猜你喜欢
阅读与作文(小学高年级版)(2021年6期)2021-09-10
大学化学(2021年7期)2021-08-29
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
通信技术(2019年8期)2019-09-03
国际呼吸杂志(2019年4期)2019-03-12
阅读与作文(小学高年级版)(2017年6期)2017-07-03
阅读与作文(小学高年级版)(2016年9期)2016-09-20
阅读与作文(小学高年级版)(2016年3期)2016-03-08
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
