
第三代EGFR-TKI耐药性机制及联合用药治疗的策略
2024-02-28 04:11周建宇秦晓红米立志
中国生物化学与分子生物学报 2024年2期
周建宇, 秦晓红, 米立志
(天津大学生命科学学院结构与分子生物学系, 天津 300072)
目前,癌症的发病率和死亡率居高不下,严重威胁着人类的健康。其中,肺癌的发病率和死亡率均居前列,是导致死亡的主要癌症类型之一[1]。如何有效治疗肺癌是近些年备受关注的热点问题。肺癌主要可分为非小细胞肺癌(non-small cell lung carcinoma,NSCLC)和小细胞肺癌(small cell lung carcinoma,SCLC)。其中,非小细胞肺癌患者比例占八成以上[2]。而表皮生长因子受体(epidermal growth factor receptor,EGFR)是非小细胞肺癌中最常见的致癌驱动基因之一,其下游信号在癌细胞增殖、分化和迁移等生理过程中发挥重要作用[3]。因此,靶向EGFR酪氨酸激酶小分子抑制剂(tyrosine kinase inhibitors,TKIs)成为治疗晚期非小细胞肺癌的最有效的靶向药物之一。目前,有三代EGFR- TKI被批准用于治疗携带EGFR突变的非小细胞肺癌患者,在使用EGFR-TKI治疗非小细胞肺癌的早期,均会取得较好的疗效。吉非替尼(Gefitinib)和阿法替尼(Afatinib)等第一代、第二代EGFR-TKI在治疗携带EGFR激活突变的晚期非小细胞肺癌患者中均取得了较好的疗效,客观应答率(objective response rate,ORR)和无进展生存期(progression-free survival,PFS)分别达到60%~70%和9~15个月,但最终仍会出现耐药现象[4]。其主要的耐药机制是由EGFR T790M突变引起,这一突变并未阻碍TKI与EGFR的结合,但却增加了对ATP的亲和力[5],从而产生耐药性,降低了第一代、第二代EGFR-TKI的治疗效果。以奥希替尼(Osimertinib)为代表的第三代EGFR-TKI为解决EGFR T790M介导的耐药问题,通过和EGFR激酶上的C797残基形成共价键,不可逆地抑制EGFR激酶活性,克服了T790M突变造成的耐药问题[6]。但服用第三代EGFR-TKI治疗的患者,在治疗数月后仍会产生耐药性。针对这一耐药性问题,本文总结与回顾了EGFR的结构、第三代EGFR-TKI药物的作用机制、已知的耐药机制、以及耐药后的治疗策略,进而我们提出若干克服EGFR-TKI靶向耐药性的可能思路。
1 表皮生长因子受体与非小细胞肺癌
EGFR是一种酪氨酸激酶,参与细胞增殖、分化以及癌症的发展[7],其突变在EGFR-TKI靶向药物的敏感性及耐药性中发挥着关键作用[8]。EGFR也称HER1或erbB1,与人类表皮生长因子受体-2(Human epidermal growth factor receptor-2,HER2)、人类表皮生长因子受体-3(Human epidermal growth factor receptor-3,HER3)及人类表皮生长因子受体-4(Human epidermal growth factor receptor-4,HER4)同属表皮生长因子受体家族[9]。如Fig.1A所示,EGFR由胞外配体结合域、跨膜域、膜旁区、胞内受体激酶区和柔性C-末端组成。当受体的胞外域结合配体后,胞外亚结构域Ⅱ、Ⅳ之间的分子内相互作用被破坏,引起受体二聚化,进而激活胞内域的酪氨酸激酶。活化的激酶通过自我/相互磷酸化C-末端,进而招募细胞内的适配分子,从而将胞外信号传递到胞内的Ras/MAPK, PI3K/Akt 或JAK/ STAT3等信号通路(Fig.1B),促进细胞的生长与繁殖[10]。正常细胞内存在一系列负反馈机制, 例如受体的去磷酸化、近膜区丝氨酸/苏氨酸残基的磷酸化、受体的内吞、循环与降解等途径, 最终将生长信号衰减和抑制[11]。
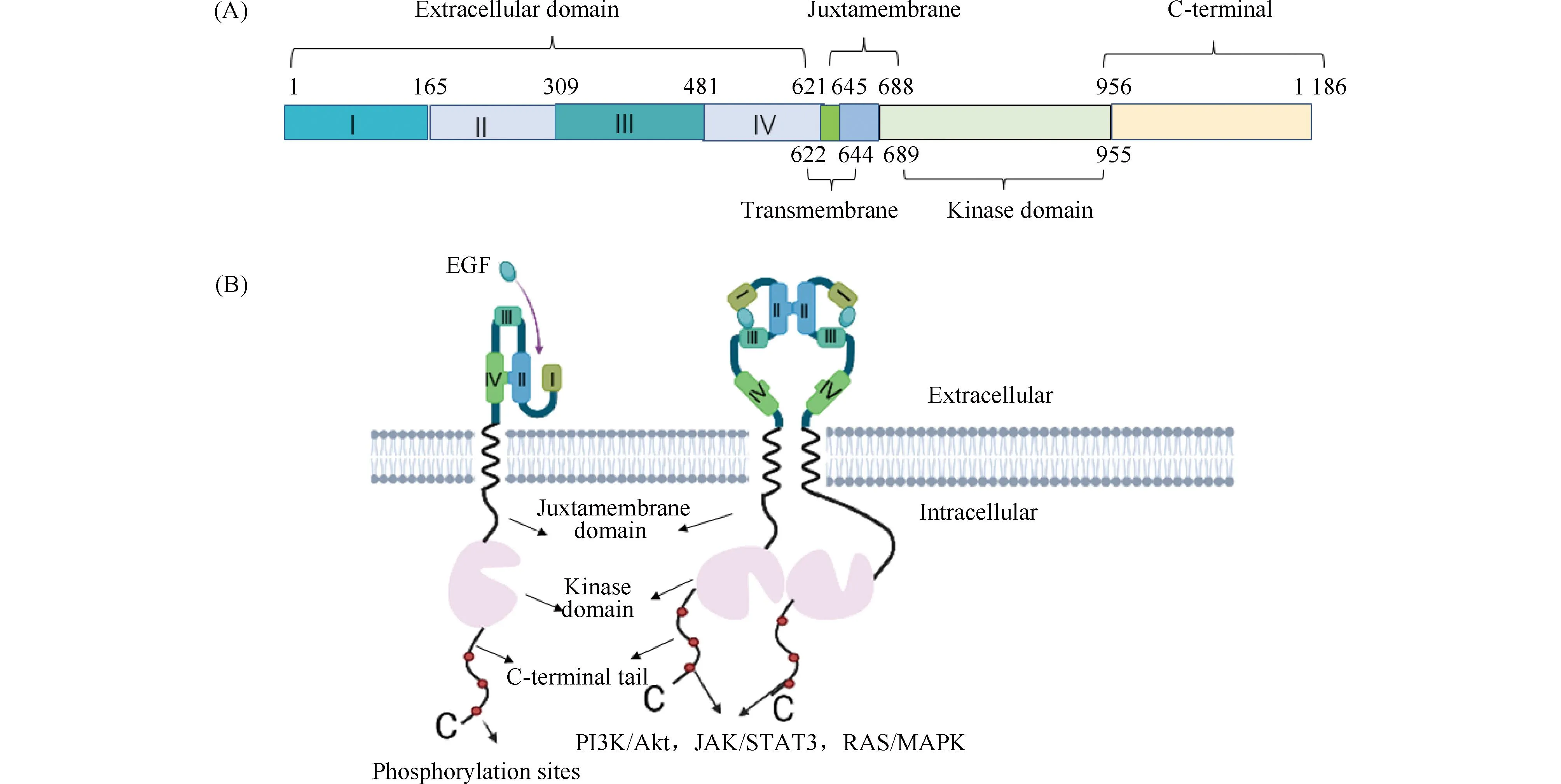
Fig.1 EGFR primary structure and receptor activation model (A) EGFR is mainly composed of extracellular domain, transmembrane domain, proximal domain, kinase domain, and C-terminal region; (B) Under the action of EGF ligands, EGFR monomers disrupt intramolecular interactions to form dimers that activate downstream signals
EGFR的过表达或异常激活是导致人类癌症发生的重要因素之一[12]。EGFR在大多数实体肿瘤中过表达,例如乳腺癌、结肠癌、头颈部、肾癌、卵巢癌和非小细胞肺癌[13],从而导致了肿瘤细胞的转移、侵袭等现象的发生。而EGFR-TKI能够选择性结合携带激活突变的EGFR激酶区,竞争性抑制EGFR与ATP的结合,阻断该信号通路,最终抑制癌细胞生长和转移,并诱导癌细胞凋亡。但由于继发突变等原因,接受靶向治疗的患者会对EGFR-TKI产生耐药性。
2 第三代EGFR-TKI的研发历程
为了解决第一代、第二代EGFR-TKI治疗中患者因携带EGFR T790M突变产生靶向耐药的问题,第三代EGFR-TKI被相继开发并应用于临床。目前,Osimertinib已成为携带EGFR突变的非小细胞肺癌患者一线治疗的标准治疗药物[14],其他在国内上市的第三代EGFR-TKI主要包括阿美替尼( aumolertinib) 和伏美替尼( furmonertinib)。这些第三代药物不可逆地结合EGFR C797,对于EGFR T790M突变具有耐受性[15]。
第三代EGFR-TKI具有相同的母核结构(Fig.2),其中Osimertinib是一种单苯胺基嘧啶化合物,具有 Michael 受体结构,能够通过不饱和丙烯酰链与EGFR ATP 结合位点的 Cys797 残基形成共价键,不可逆地结合在EGFR激酶的活性中心,抑制激酶活性,阻断其下游信号[16]。Osimertinib 对携带 EGFR激活突变和 T790M 耐药突变的肿瘤细胞具有选择性抑制功能。Aumolertinib和 Furmonertinib是在Osimertinib的结构基础上进行优化的。Aumolertinib采用环丙基替换Osimertinib吲哚环上的甲基,这样能够和 Met790 侧链形成疏水相互作用,从而提高对EGFR T790M突变型肿瘤细胞的抑制作用和选择特异性。同时环丙基脂溶性高,使药物对血脑屏障的穿透能力强。而Furmonertinib则是在母核结构上引入N原子,同时用三氟乙基和吡啶环分别取代Osimertinib上的甲基和苯环,以防止非选择性代谢产物的产生[17],提高药物活性和激酶选择特异性,降低了脱靶效应,显著地提高了药物的疗效。总的来说,第三代EGFR-TKI具有更高的选择特异性和更低的毒性[18],在临床上应用效果更佳。
3 表皮生长因子受体依赖的第三代EGFR-TKI耐药
目前,已知的第三代EGFR-TKI耐药机制可分为EGFR依赖型和非EGFR依赖型(Fig.3)。
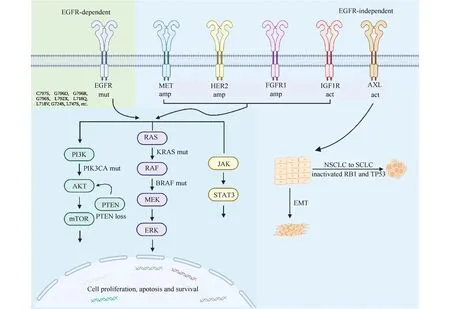
Fig.3 Classification of main drug resistance mechanisms of the third generation EGFR-TKI amp: amplification; mut: mutation. Mainly including EGFR mutation, abnormal activation of bypass and downstream signals and histological phenotypic transformation[19]
3.1 EGFR C797S突变
EGFRC797S突变直接导致了Osimertinib无法与EGFR激酶共价结合,致使其因丧失对EGFR激酶的高亲和力而失效。Osimertinib用于一线治疗时,出现耐药性的患者中有约7%的患者具有C797S突变[20],而在Osimertinib用于二线治疗时,耐药患者中出现C797S突变的频率达到14%,居二线治疗中所有耐药机制发生频率的首位,可见Osimertinib在一线和二线治疗非小细胞肺癌中的耐药机制存在差异[21]。有数据表明,二线治疗时,携带C797S突变的患者通常也会保留T790M突变,而C797S突变和保留的T790M突变又可根据二者是否位于同一条DNA链上分为顺式结构和反式结构(Fig.4)[22]。当EGFR C797S和T790M的突变为反式结构时(位于不同等位基因),肿瘤细胞依旧保留对第一代、第二代EGFR TKI敏感性,联合第三代EGFR-TKI可有效抑制肿瘤细胞的生长;但是对于顺式结构的突变目前仍无有效的用药策略[23]。

Fig.4 Diagram of the cis and trans structures of the EGFR T790M and C797S mutation at the allele The T790M and C797S mutations are cis structures located on the same DNA strands; The T790M and C797S mutations are trans structures located on different DNA strands
3.2 其他EGFR罕见突变
在NCLSC晚期,患者体内还能够检测到耐药相关的罕见EGFR突变,例如G796D、G796R[24]、G796S[25]、L792X、L718Q[26]、L718V[27]、G724S[28]和L747S[29]等。与C797相邻的G796发生G796S、G796R、G796D等突变均为溶剂前沿突变,对于Osimertinib与EGFR的结合具有空间位阻效应,其中G796R突变的影响最大[30, 31]。EGFR L792X突变位于EGFR蛋白激酶结构域中N-lobe与C-lobe连接的铰链区口袋,能在空间上干扰Osimertinib苯环上的甲氧基,破坏其与激酶结构域的结合[31]。EGFR L718残基位于EGFR激酶结构域的ATP结合部位,其发生L718Q和L718V突变均能够通过空间位阻来阻止EGFR-TKI与EGFR结合,在2%的Osimertinib一线治疗患者中检测到L718Q突变[32]。
4 表皮生长因子受体非依赖的第三代EGFR-TKI耐药
非EGFR依赖性耐药是指与EGFR自身突变无关的耐药机制,通常是由于出现一种或多种旁路信号激活、上调下游信号或发生组织表型转换所导致。因此,一般无法通过测序检测出这一类型的耐药。
4.1 EGFR旁路信号激活
4.1.1 HGF过表达与C-Met扩增 C肝细胞生长因子受体(cellular-Mesenchymal-epithelial transition factor,C-Met)的高表达在肺癌对EGFR‐TKIs产生耐药性的过程中发挥重要作用[33]。携带EGFR激活突变的非小细胞肺癌患者常伴有C-Met扩增[34]。而肝细胞生长因子(hepatocyte growth factor,HGF)是C‐Met的配体,可通过激活C-Met通路,诱导癌细胞对EGFR‐TKI产生抵抗[35, 36]。携带野生型EGFR肿瘤患者血浆中HGF水平显著升高。但无论EGFR突变状态如何,低HGF水平与EGFR-TKI较高的应答率、较长的无进展生存期(PFS)和总生存期(OS)显著相关。在对FLAURA的研究数据进行多变量分析时发现,EGFR突变(P=0.002)和低HGF水平(P=0.031)均可独立预测PFS的延长,而活动状态(performance status,PS)为0(P=0.001)和 成纤维细胞生长因子(fibroblast growth factor, FGF)低水平(P=0.002)则可独立预测OS的延长[36]。
4.1.2 HER2扩增 HER2与EGFR同属EGFR受体酪氨酸激酶家族。它会激活PI3K/Akt等下游通路,其扩增在众多EGFR-TKI耐药性机制中占有相当大的比例[37]。在Osimertinib用于一线治疗所产生耐药的患者中,发现大约2%的患者有HER2的扩增,在二线治疗和临床筛查中也发现有HER2扩增[20]。体外实验表明,EGFR T790M突变细胞系中过表达HER2能够降低该细胞系对Osimertinib和罗西替尼(Rociletinib)的敏感性,使其对2种TKI药物产生耐药性[38]。
4.1.3 IGF1R异常 1型IGF受体(Insulin-like growth factor 1 receptor, IGF1R)是一种跨膜酪氨酸激酶受体,可以被胰岛素样生长因子(IGF1或IGF2)激活,引起自身酪氨酸激酶结构域的磷酸化并引发胞内信号传导,调控细胞的生长和分化,参与高等生物的生长、发育、衰老等各种生命活动[39]。IGF1R的异常激活也是造成EGFR-TKI耐药的重要原因之一[40]。在携带EGFR T790M 突变的非小细胞肺癌中,IGF1R介导的信号传导在EGFR-TKI发生耐药的早期阶段发挥作用。由于IGF1R的竞争蛋白质-胰岛素样生长因子结合蛋白-3(IGFBP3)表达缺失,IGF1R信号得以激活,导致肿瘤细胞对EGFR-TKI耐药;而引入IGF1R抑制剂BMS 536924能使肿瘤细胞恢复对EGFR-TKI的敏感性[41]。
4.1.4 成纤维细胞生长因子受体信号传导 成纤维细胞生长因子受体(FGFR)是一种酪氨酸激酶受体,调节细胞增殖和分化等生物过程,同时也是癌症治疗的一个重要靶点[42]。在Osimertinib耐药的患者中发现,有FGFR1扩增及其配体FGF2的表达上调,这表明FGF2-FGFR1信号通路可能是导致Osimertinib耐药的一个潜在因素[43]。
4.2 EGFR下游信号激活
4.2.1 K-RAS突变 RAS是由原癌基因编码的一种小型GTP结合蛋白,介导膜表面生长因子受体、细胞内信号通路和转录因子活性的偶联,是EGFR重要的下游信号通路。RAS /RAF /MEK/ERK信号通路异常激活会导致对EGFR-TKI耐药[44],并与肿瘤发生、侵袭和不良预后相关[45]。在经Afatinib治疗后再使用Osimertinib治疗,一段时间后在产生耐药的患者中检测到 K-RAS突变和 EGFR 高表达[46]。由于K-RAS和GTP的高亲和力以及其独特的结构,使得开发靶向K-RAS的竞争性抑制剂充满挑战,因此,临床上急需探索全新的治疗方法来解决K-RAS突变带来的耐药问题[47, 48]。
4.2.2 PTEN缺失PTEN(Phosphatase and tensin homolog)是目前为止发现的第1个具有磷酸酶活性的抑癌基因,编码具有蛋白质磷酸酶和磷脂磷酸酶活性的双磷酸酶,在PI3K/Akt信号通路中发挥重要作用[49]。PTEN作为磷脂酰肌醇3,4,5-三磷酸(PIP3)脱磷的脂质磷酸酶,能降低PIP3的水平,负调控PI3K/AKT信号通路,从而达到抑癌目的[50]。对2名Osimertinib二线治疗中出现原发性耐药的非小细胞肺癌患者进行检测,发现了与PTEN缺失有关的K-RAS G12D突变的情况,该突变在Osimertinib一线治疗和后续治疗出现的耐药中都有报道[51]。
4.2.3 BRAF突变BRAF(v-raf murine sarcoma viral oncogene homolog B1)编码的BRAF蛋白,在RAF信号通路中发挥重要作用,是MAPK信号通路中一个非常重要的信号转导分子[52]。BRAF突变可以导致MAPK信号通路活性增强、细胞周期调控紊乱、细胞生长及迁移失控等,进而导致肿瘤的发生与发展,以及肺癌和结直肠癌等对EGFR-TKI治疗的耐药抵抗[53, 54]。在约1%的非小细胞肺癌获得性耐药患者样本中发现有BRAF突变(G469A和V600E),而BRAF突变与预后不良等密切相关[55]。研究者发现,靶向BRAF V600E 突变的抑制剂Enorafenib 与Osimertinib联合使用可有效抑制耐药细胞的生长[56]。
4.2.4 PIK3CA突变PIK3CA(Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha)是一种常见的致癌基因,编码PI3K的催化亚基,PIK3CA基因突变通过PIK3/Akt的途径引发AKT持续活化,抑制细胞凋亡,造成对 EGFK-TKI的耐药[57]。在接受一线Osimertinib治疗的患者中,在6例病例中发现了PIK3CA E453K、E545K和H1047R突变,其中E545K最具代表性(4% ),并且 PIK3CA E545K突变在介导Osimertinib耐药性中的作用已在体外得到证实[32, 58, 59]。对在Osimertinib二线治疗耐药的患者进行检测,发现有约10%的患者有PIK3CA的扩增和突变,例如E545K、E542K、R88Q、N345K和E418K等突变[21, 51, 60-62]。
4.3 组织学表型转变
4.3.1 上皮-间充质转化 上皮-间充质转化(epithelial-mesenchymal transition,EMT)是指上皮细胞转化为间充质细胞的生物学过程,主要表现为E-钙黏着蛋白(E-cadherin)表达的丧失和纤连蛋白(fibronectin)及波形蛋白(vimentin)表达的增加,是独立于T790M突变肺腺癌患者产生EGFR-TKI耐药的机制[63]。对Osimertinib获得性耐药的非小细胞癌患者的细胞表现出了上皮细胞连接蛋白质(例如E-cadherin)减少、间质细胞标记(例如vimentin、fibronectin)增加等EMT典型特征[64]。在使用抑制剂逆转EMT过程后,获得性耐药的肿瘤重新恢复了对Osimertinib的敏感性[65]。
4.3.2 细胞类型转化 这里的细胞类型转化特指非小细胞肺癌发生靶向耐药后转化为小细胞肺癌的过程,但转化后的肿瘤细胞仍携带EGFR致癌驱动突变,即小细胞肺癌系由EGFR突变进化而来。2006年,一名45岁的女性非小细胞肺癌患者在接受Erlotinib治疗后,在2次活检中首次发现小细胞肺癌的组织类型转化[66]。在Osimertinib一线及二线治疗后出现耐药情况的患者中,分别有14% 及4~15%发生了由EGFR突变的非小细胞肺癌向小细胞肺癌的组织学转变[60, 67]。值得注意的是,非小细胞肺癌转化为小细胞肺癌的风险与视网膜母细胞瘤1(Retinoblastoma1, Rb1)和p53肿瘤蛋白 (Tumor protein 53, TP53)的突变显著相关,在携带野生型RB1和TP53的患者中暂时未发现小细胞肺癌转化病例[68],因此,RB1和TP53的突变可能是这种细胞类型转化的生物标志之一。
除上述耐药机制外,ABC转运蛋白(ATP-binding cassette transporter,ABC)介导的小分子药物外排同样在EGFR-TKI耐药中发挥重要作用。ABC转运蛋白是一种以ATP(三磷酸腺苷)为驱动能源的跨膜运输蛋白质,能够将小分子药物从细胞中转运出来,降低癌细胞中的药物积累浓度[69]。
5 克服第三代EGFR-TKI获得性耐药的治疗策略
5.1 开发第四代EGFR-TKI
2016年,报道了第1个靶向EGFR酪氨酸激酶的变构抑制剂EAI045[70],它通过与激酶变构位置结合,以改变其构象的方式抑制酶促反应速率。在小鼠模型中,它与 EGFR 单抗药物西妥昔联合使用能显著抑制携带EGFR L858R/T790M/C797S 突变的肿瘤生长,但遗憾的是该化合物单独使用未能展现应有的疗效,进入临床使用的前景堪忧[58, 70, 71]。2017年,报道的EGFR别构抑制剂布加替尼(Brigatinib)是由一组以 U 型构象围绕双苯胺嘧啶支架构建的二甲氧基[72]。Brigatinib和 EGFR 单抗药物(例如帕尼单抗或西妥昔单抗)联用,能够克服 C797S突变导致的第三代EGFR-TKI耐药,在小鼠模型中显示出良好的抗肿瘤效果。在2020年,公布了一款名为CH723316的第四代EGFR-TKI抑制剂,体外小鼠实验结果显示,CH7233163对EGFR三基因突变和EGFR单、双活化突变也取得很好的疗效。在CH7233163应用到Del19/L858R/T790M、L858R/T790M突变和Del19的小鼠体内,均可明显观察到肿瘤体积的缩小,CH7233163彰显了良好的临床应用前景[73]。目前,仍然未见第四代EGFR-TKI批准用于临床,其开发与应用仍然任重而道远。
5.2 联合治疗策略
5.2.1 与其他靶向的小分子抑制剂联合使用 目前,因第三代EGFR-TKI耐药机制多样,联合使用不同的小分子抑制剂是克服靶向耐药的策略之一。针对EGFR C797S突变所造成的三代EGFR-TKI不同的耐药性问题,可以通过测序确定其与T790M是否为反式结构,如是,则可将第一代和第三代EGFR-TKI联合使用抑制耐药肿瘤细胞的生长[22]。针对C-Met高表达引发的对Osimertinib、Rociletinib等EGFR-TKI耐药的问题,联合使用C-Met抑制剂能增强肿瘤细胞对Osimertinib的敏感性[74]。Osimertinib和ALX抑制剂cabozantinib联合使用能够显著抑制ALX高表达,并携带EGFR T790M突变的耐药肿瘤细胞在体内外的生长[75]。Osimertinib联合MEK抑制剂司美替尼(Selumetinib)能够抑制由KRAS过表达引起的耐药细胞系的生长和增殖,而Osimertinib联合另一种MEK抑制剂曲美替尼(Trametinib)可以克服KRAS突变引起的耐药[46]。此外,三代EGFR-TKI联合使用氧化磷酸化(OxPhos)抑制剂、HER2抑制剂等在治疗特定类型肿瘤耐药均有很好的疗效[76, 77]。
5.2.2 与单克隆抗体联合使用 目前,临床上使用的EGFR抗体药物有西妥昔单抗、panitumumab、耐昔妥珠单抗(necitumumab)以及尼妥珠单抗(nimotuzumab),它们皆通过与EGFR胞外域结合来阻断EGFR与其配体的识别或受体二聚化。第四代EGFR抑制剂布甲替尼和西妥昔单抗的联合靶向治疗,能够改善获得EGFR T790M-cis-C797S突变引起的Osimertinib耐药,延长患者的生存期,这也证明了EGFR-TKI联合单克隆抗体治疗可能是一种有效克服EGFR-TKI耐药的治疗策略[78]。西妥昔单抗和曲妥珠单抗的三联体(3×mAb)可抑制携带C797S突变的肿瘤,诱导肿瘤细胞衰老,并且在与Osimertinib联合治疗中展现出协同效应,可持续抑制肿瘤[79]。
5.2.3 与化疗联合使用 化疗是治疗各类癌症的一种普遍采用且非常重要的手段,也是EGFR-TKI靶向耐药后经常采用的治疗策略。例如,在治疗小细胞肺癌转化引起的EGFR-TKI耐药中,在患者产生对Osimertinib耐药后使用铂类双联化疗仍取得了一定疗效,但最终未能阻止癌症的转移[80]。
5.2.4 与免疫检查点抑制剂联合使用 以免疫检查点抑制剂(immune checkpoint inhibitors,ICIs)为代表的免疫疗法在肿瘤治疗中取得重大突破。这些免疫检查点抑制剂,包括PD-1/PD-L1抑制剂和CTLA-4抑制剂等,通过调节肿瘤患者自身的免疫反应达到杀伤肿瘤的目的[81]。但多项临床试验显示,针对携带EGFR激活突变的肺癌患者既不适合单独使用免疫检查点抑制剂,也不适合与EGFR-TKI联用,两者均无法取得明显治疗效果[82]。但针对携带鼠类肉瘤病毒癌基因(kirsten rat sarcoma viral oncogene, KRAS)与TP53共突变的患者,靶向治疗与免疫检查点抑制剂联用效果较好,可能与携带KRAS突变的患者肿瘤突变负荷(tumor mutational burden, TMB)较高有关。但对于携带其他主要致癌驱动基因突变(例如EGFR、间变性淋巴瘤激酶(anaplastic lymphoma kinase , ALK)等)的患者,其TMB相对较低[82-83]。免疫检查点抑制剂联合TKI疗效不明确,且存在安全性隐患,尚需进一步探索适用人群与适应症。
目前,多种药物联合应用对第三代EGFR-TKI治疗后耐药问题的解决展现了良好的希望[84]。然而,组合多种EGFR-TKI进行联合应用治疗时,如何管理这些分子之间的直接相互作用以及患者联合治疗引起的毒性是目前的最大问题。
6 问题与展望
EGFR的功能异常在非小细胞肺癌发生、发展中发挥至关重要的作用,EGFR-TKI靶向治疗成为当下治疗晚期非小细胞肺癌的一线疗法之一。相对于化疗,EGFR-TKI在治疗药物敏感人群时展现出良好安全性,并能够显著延长晚期非小细胞肺癌患者的生存期。但是由于EGFR突变肿瘤中第三代EGFR-TKI耐药性的机制在患者之间是不同的,并且在不同的肿瘤部位之间存在异质性问题,第三代EGFR-TKI的长期使用仍然受限于耐药性的产生。相较于前两代药物,由于EGFR T790M突变产生的耐药机制,第三代EGFR-TKI的耐药机制更加复杂多变,因此,对其血液进行液体活检或组织样本进行高通量测序,以阐明耐药性机制,对指导下一步治疗具有重要意义,这将有助于克服耐药性的新型联合疗法的临床研究。虽然目前已经发现多种不同的耐药机制,但是仍有大量病例存在未知的耐药机制。目前,开发第四代EGFR-TKI及探索不同的联合治疗策略,以期克服第三代EGFR-TKI的耐药问题,也是基础研究和临床医学所面临的重大挑战。阐明新的三代EGFR-TKI耐药机制,及时监测、发现治疗中产生的获得性耐药,开发新的联合治疗策略,可能是未来克服EGFR-TKI靶向耐药,提高患者生存期的有效手段。
猜你喜欢
保健医苑(2022年5期)2022-06-10
中国临床医学影像杂志(2021年6期)2021-08-14
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
肝博士(2020年5期)2021-01-18
中国医药生物技术(2015年4期)2015-12-26
中国医药生物技术(2015年4期)2015-12-26
中国现代医学杂志(2015年26期)2015-12-23
医学研究杂志(2015年7期)2015-06-22
西南国防医药(2015年11期)2015-02-28
