
缺血性脑卒中后促进转录因子EB核转位改善神经元自噬流障碍的分子机制
2024-02-28 04:11李尚丹邓仪昊何红云
中国生物化学与分子生物学报 2024年2期
李尚丹, 邓仪昊, 何红云
(1)昆明理工大学基础医学院人体解剖学教研室, 昆明 650500;2)昆明理工大学附属安宁市第一人民医院医院办公室, 昆明 650399)
缺血性脑卒中 (ischemic stroke,IS)是一种因大脑局部血流中断造成中枢神经系统 (central nervous system,CNS)损伤的脑功能障碍疾病[1],诱发兴奋性毒性、氧化应激、神经炎症、细胞凋亡等一系列级联反应,进一步加重缺血区神经元损伤甚至死亡[2]。在缺血性脑卒中缺血、缺氧和营养缺乏等应激条件的刺激下,自噬被不同程度激活以减轻神经元损伤[3]。自噬是一种由溶酶体介导的细胞内分解代谢机制,蛋白质、受损线粒体和内质网等自噬底物被膜包裹成自噬体,随后与溶酶体融合,利用溶酶体中的酶将自噬底物降解后再利用,以维持细胞正常的结构和功能[4],这一动态的连续变化过程称为“自噬流”[5]。
自噬流中任何一个步骤的中断都会导致自噬流障碍[6]。自噬流障碍会导致自噬底物无法被及时降解进行再利用,进而加重大脑缺血区神经元损伤[7]。其中,溶酶体功能障碍和自噬体-溶酶体融合障碍是导致自噬流障碍的主要因素(Fig.1)[6]。溶酶体功能和自噬体-溶酶体融合严格受到转录因子EB(transcription factor EB,TFEB)的调控[8]。TFEB的活性与其磷酸化状态有关[9]。当TFEB的2个特殊的丝氨酸残基Ser142和Ser211均被磷酸化时,TFEB留存在细胞质中为无活性状态[10]。而去磷酸化的TFEB具有活性,会转位进入细胞核激活自噬相关基因的表达,增强溶酶体功能并促进自噬体-溶酶体融合,进而改善自噬流障碍,减轻缺血性脑卒中后的神经元损伤[11]。

Fig.1 Neuronal autophagic flux dysfunction in ischemic stroke (Created with MedPeer (www.medpeer.cn)) The substrates to be degraded are engulfed by a flat membrane called an isolated membrane. When it is wrapped into a closed double-membrane organelle, autophagosomes are formed. Mature autophagosomes fuse with lysosomes to form autolysosomes. The components to be degraded are finally hydrolyzed by various acidic hydrolases in lysosomes, and the degraded components are recycled by cells. This continuous process is called autophagic flux. The interruption of any step in the autophagic flux of ischemic stroke will lead to autophagic flux dysfunction. The proteins and cellular components in the cells cannot be cleared in time and accumulate in large quantities, which in turn aggravates neuronal damage in the cerebral ischemic area
本文将详细阐述缺血性脑卒中后TFEB核转位的分子机制,进一步分析TFEB核转位可增强溶酶体功能并促进自噬体-溶酶体融合,以期为未来临床研究靶向TFEB改善自噬流障碍治疗缺血性脑卒中提供理论依据。
1 自噬流障碍是脑缺血后神经元损伤的重要病理机制
溶酶体功能受损、过度自噬或自噬不足、自噬体-溶酶体融合障碍等都会导致自噬流障碍,使细胞中蛋白质和细胞成分无法被及时清除而大量聚集,诱发多种人类疾病,包括神经系统疾病、心血管疾病和癌症等[12]。溶酶体发挥降解作用是依靠其内部的酸性水解酶实现的,包括组织蛋白酶D (cathepsin D,CTSD)、组织蛋白酶B (cathepsin B,CTSB)、组织蛋白酶L (cathepsin L,CTSL)等[13]。其中,CTSD是参与自噬-溶酶体降解底物的主要水解酶,改变CTSD活性或表达水平都可能会导致溶酶体功能受损[14]。Hossain[15]等研究发现,在原代神经元细胞中敲除CTSD会导致溶酶体功能障碍,进一步加重大脑缺血性神经元损伤。因此,溶酶体功能受损会引发自噬流障碍,加快缺血性脑卒中的病理进程。
在脑缺血时,过度自噬或自噬不足也会诱发细胞死亡并加重缺血性脑损伤[4]。过度自噬使细胞成分被溶酶体过度降解,造成降解产物大量堆积,引发自噬流障碍,导致细胞死亡[16]。在缺血性中风小鼠中,白细胞介素 (interleukin,IL)-17A水平显著升高,IL-17A介导的过度自噬会加快神经元凋亡和坏死,从而加重缺血性脑损伤[17]。而自噬水平不足使得自噬底物不能被溶酶体有效降解,引发底物堆积,也会导致自噬流障碍[16]。此外,小鼠脑缺血会诱导miR-497表达,miR-497过表达会降低自噬水平,使自噬水平不足,诱导缺血后神经元的死亡[18]。由此可见,过度自噬或自噬不足都会引起自噬流障碍,继而加重缺血性脑损伤。
自噬体与溶酶体的融合是自噬完成的关键,自噬体-溶酶体融合障碍会导致自噬-溶酶体不能形成,阻断自噬流[19]。既往研究表明,突触融合蛋白17(syntaxin 17,STX17)是参与自噬体和溶酶体融合的重要可溶性N-乙基马来酰亚胺敏感因子附着蛋白受体(soluble N-ethylmaleimide-sensitive factor attachment protein receptor,SNARE)蛋白,使用腺相关病毒9 (adeno-associated virus 9,AAV9)敲低小鼠大脑中STX17会抑制自噬体和溶酶体的融合,加重脑缺血再灌注损伤 (ischemia-reperfusion injury,IR)程度[20]。因此,自噬体与溶酶体融合障碍会导致神经元自噬流障碍,加剧脑缺血诱导的神经元损伤。
综上所述,自噬流障碍是脑缺血后神经元损伤的重要病理机制。新近研究表明,TFEB核转位会通过及时清除自噬底物以促进细胞更新,改善自噬流障碍,对神经元细胞发挥保护作用[21]。
2 转录因子EB核转位可改善卒中后的自噬流障碍
TFEB是进化上保守的小眼畸形转录因子 (microphthalmia associated transcription factor,MITF)家族成员之一,具有典型的碱性螺旋-环-螺旋-亮氨酸-拉链 (basic helix-loop-helix leucine zipper,bHLH-Zip)结构[22]。TFEB活化会激活自噬,进而调控凋亡、炎症和氧化应激等信号过程[23]。更具体地说,TFEB的激活会促进自噬流,刺激线粒体自噬,限制活性氧 (reactive oxygen species,ROS)的产生,抑制炎症小体的激活,并抑制白介素的分泌,最终抑制细胞焦亡和炎症等反应[24, 25]。
TFEB是自噬流信号通路的关键中枢调节因子。TFEB与位于自噬和溶酶体基因启动子区域的“协同溶酶体表达与调控 (coordinated lysosomal expression and regulation, CLEAR)”元件结合,进而驱动自噬过程和溶酶体生物发生相关基因的表达,包括自噬启动相关基因 (BECN1、WIPI1、ATG9B和NRBF2)以及自噬体与溶酶体融合相关基因 (UVRAG,RAB7)等基因[26]。TFEB核转位通过促进自噬体形成和自噬体-溶酶体融合,进而加快清除自噬底物,利于神经元存活[27]。根据研究显示,在大鼠大脑皮质内源性神经干细胞中敲除TFEB会导致自噬流信号通路受损,使胞内蛋白质聚集体和受损细胞器无法被及时清除而积累,对神经系统造成损伤[28]。此外,拟人参皂苷F11 (pseudoginsenoside F11,PF11)在永久性脑缺血期间通过上调钙调磷酸酶(calcineurin,CaN)活性进而促TFEB核转位,减少自噬体及底物的异常堆积,以减轻自噬流障碍和缺血诱导的神经元损伤[29]。
综上,TFEB是自噬流信号通路的关键调节因子,TFEB核转位可改善卒中后自噬流障碍。而TFEB的表达、定位和活性受到翻译后修饰的严格调控[30]。
3 转录因子EB核转位的调控机制
TFEB的细胞定位和活性主要由其磷酸化调控[31]。生理条件下,磷酸化的TFEB与14-3-3黏附蛋白结合形成非活性形式的复合物锚定在细胞质中[32]。当细胞受到饥饿、溶酶体功能异常等外界刺激时,去磷酸化的TFEB与14-3-3黏附蛋白解离后活化,随后从细胞质转移到细胞核(Fig.2)[10]。
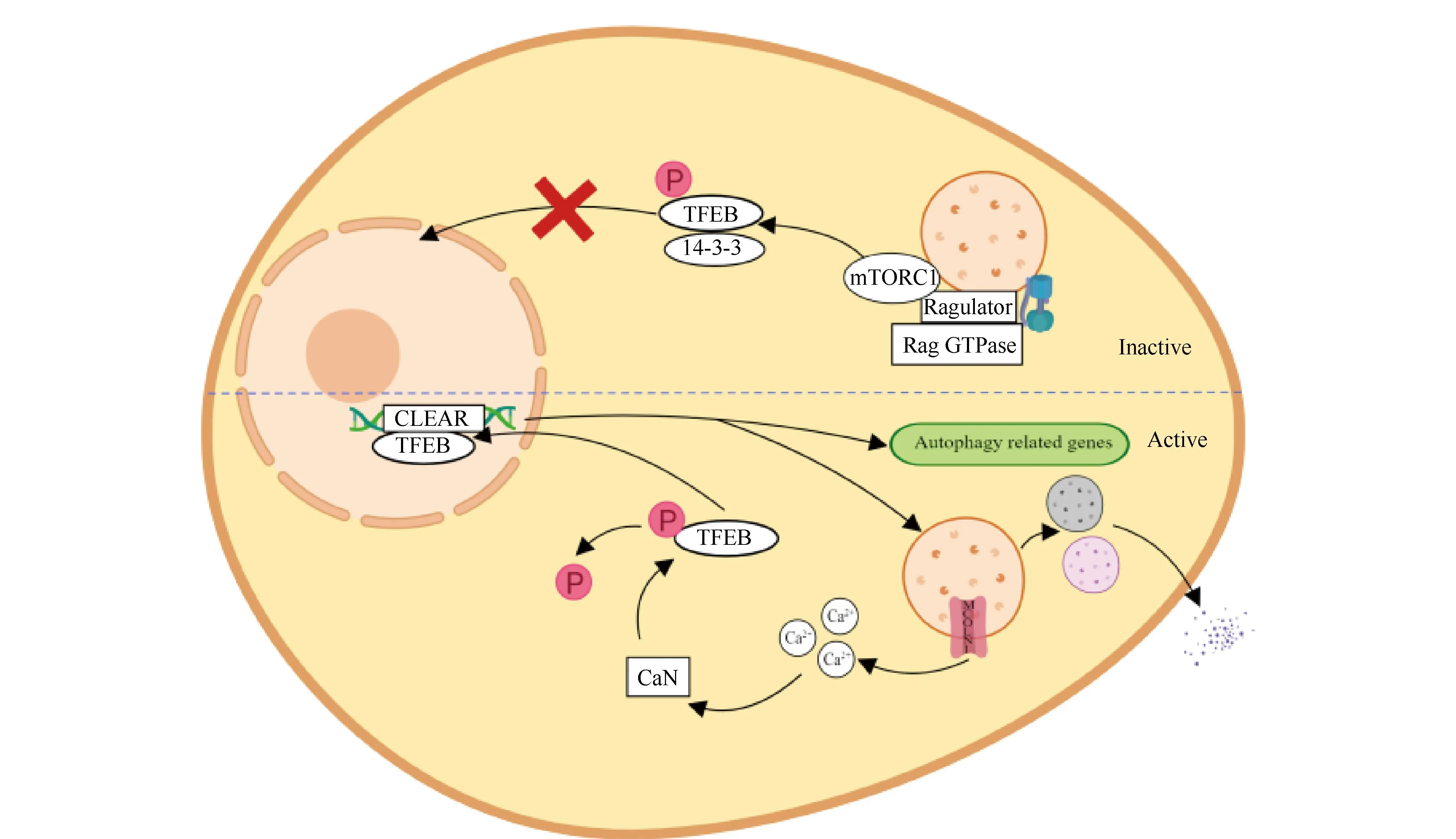
Fig.2 TFEB nuclear translocation enhances autophagic flux (Created with MedPeer (www.medpeer.cn)) The cellular localization and activity of TFEB are closely related to its phosphorylation. The activation of mTORC1 can inhibit its nuclear translocation by phosphorylating TFEB. Calcineurin mediates the dephosphorylation of TFEB.Dephosphorylated TFEB enters the nucleus and binds to the CLEAR sequence in the nucleus, promotes the expression of downstream lysosomal genes, and enhances the biosynthesis and physiological function of lysosomes
TFEB磷酸化由多种激酶催化,使其保留在细胞质中,包括哺乳动物雷帕霉素靶蛋白1 (mammalian/mechanistic targets of rapamycin complex 1,mTORC1)、蛋白激酶C-β (protein kinase C-beta,PKC-β)、细胞外信号调节激酶 (extracellular signal regulated kinase,ERK)、蛋白激酶B (protein kinase B,Akt)、糖原合酶激酶-3β (glycogen synthase kinase-3beta,GSK-3β)等[10]。其中,mTORC1介导的TFEB磷酸化是最广泛研究的信号转导途径[31]。TFEB是mTORC1的直接下游靶标[33]。在营养丰富、无溶酶体应激和液泡型三磷酸腺苷酶 (vacuolar-type adenosinetriphosphatases,V-ATPases)不被抑制的条件下,V-ATPases、Ragulator复合体和Rag GTP酶形成活性复合物,在溶酶体表面与mTORC1结合并激活mTCOR1[34]。mTORC1介导TFEB的2个丝氨酸残基Ser142和S211磷酸化,磷酸化的TFEB与细胞质中的14-3-3黏附蛋白结合,掩盖位于Ser241和Ser252之间的核定位信号 (nuclear localization signal,NLS),将TFEB保留在细胞质中[35, 36]。而在饥饿、溶酶体应激或V-ATPases抑制的条件下,Rag GTP酶被关闭,其从溶酶体表面释放mTORC1并将其灭活[22]。由于mTORC1不能再磷酸化TFEB,去磷酸化的TFEB转位到细胞核中并与其靶基因的CLEAR序列结合,上调自噬和溶酶体基因的表达[37]。
钙调磷酸酶使TFEB去磷酸化,诱导TFEB由细胞质转位到细胞核,激活其转录活性,以诱导激活自噬和溶酶体相关靶基因的转录[38]。在饥饿或高能耗的情况下,溶酶体通过粘脂蛋白1 (mucolipin 1,MCOLN1)释放钙离子,引起局部钙信号转导,激活钙调磷酸酶,钙调磷酸酶结合TFEB并使其去磷酸化,从而促进TFEB核转位和靶基因转录[39]。
由此可见,TFEB通过磷酸化、去磷酸化在细胞质和细胞核之间不断穿梭来快速响应环境变化,从而维持细胞稳态。
4 转录因子EB核转位改善自噬流障碍减轻缺血性脑损伤的机制
4.1 TFEB核转位增强溶酶体功能高效代谢底物
溶酶体在调节自噬流信号通路中发挥关键作用,是参与细胞内底物降解过程的细胞器,维持其功能的正常是维持细胞稳态所必需的[40]。TFEB磷酸化会抑制其核转位,使溶酶体生物合成减少,最终导致溶酶体功能障碍[41]。而TFEB去磷酸化激活核转位时,粘脂蛋白1(mucolipin 1,MCOLN1)介导溶酶体释放钙离子,调节溶酶体胞吐作用及溶酶体酶的分解代谢活性,以增加溶酶体的数量并增强溶酶体功能,加快病理条件下的细胞降解清除底物的速率[42]。我们以往的研究发现,脑缺血发生时,GSK-3β会增加TFEB磷酸化,抑制TFEB核转位,使溶酶体生物合成减少,引起溶酶体功能障碍,加重神经元损伤。而抑制GSK-3β能促进神经元中TFEB核表达,增强神经元溶酶体功能,以达到神经保护的目的[43]。因此,TFEB核转位增强溶酶体的功能后会高效降解代谢底物,进而改善自噬流障碍,从而发挥对脑缺血损伤的神经保护作用。
4.2 TFEB核转位介导自噬体与溶酶体融合上调自噬流
自噬体和溶酶体的融合是溶酶体降解自噬底物的基础[44]。TFEB核转位不仅增强溶酶体功能,而且促进自噬体-溶酶体融合[45]。新近研究表明,运动促进皮层中TFEB的核转位,促进自噬体与溶酶体融合,形成自噬-溶酶体,上调自噬流活性,有助于提高突变蛋白的清除效率[46]。并且,海藻糖通过上调腺苷5′-单磷酸激活蛋白激酶和辅激活蛋白关联精氨酸甲基转移酶1活性,进而诱导TFEB激活,促进自噬体与溶酶体融合,改善自噬流障碍,有效降低高盐喂养的卒中型自发性高血压大鼠的卒中发病率[47]。此外,蜜二糖通过促进神经元中的TFEB核转位促自噬-溶酶体形成,改善自噬流障碍,从而赋予对脑缺血再灌注损伤的神经保护作用[48]。上述结果表明,TFEB核转位调节自噬相关基因的表达,促进自噬体与溶酶体融合,改善缺血性脑卒中后自噬流障碍。
5 问题与展望
自噬流障碍与缺血性脑卒中的发病机制密切相关,而TFEB在自噬流信号通路中发挥关键调节作用。目前,通过靶向TFEB以调节自噬流信号通路是减轻缺血后脑损伤程度的有效方法,但由于TFEB下游机制、靶基因以及TFEB激活的潜在机制尚未完全确定,促进TFEB核转位的方法和分子机制仍有待进一步研究探索。此外,靶向TFEB调控自噬途径的生物活性物质尚处于起步阶段,未来具有巨大潜力。加大对TFEB的分子机制和药物的研究力度有望成为开发治疗缺血性脑卒中的新策略。
猜你喜欢
云南化工(2021年6期)2021-12-21
生物化工(2021年2期)2021-01-19
科学(2020年2期)2020-08-24
生物化工(2020年1期)2020-02-17
读与写(2019年35期)2019-11-05
现代职业教育·高职高专(2018年7期)2018-05-14
中国康复理论与实践(2015年10期)2015-12-24
吉林大学学报(医学版)(2015年5期)2015-12-16
中国体外循环杂志(2015年3期)2015-12-08
生物技术通报(2015年1期)2015-04-10
